|
|
|
RESOLUCIÓN 3157 DE 2018
(julio 26)
Diario Oficial No. 50.667 de 27 de julio de 2018
MINISTERIO DE SALUD Y PROTECCIÓN SOCIAL
Por la cual se expide la “Guía para el desarrollo y presentación de los estudios de estabilidad de medicamentos de síntesis química”.
EL MINISTRO DE SALUD Y PROTECCIÓN SOCIAL,
en ejercicio de sus facultades legales, en especial, las conferidas por el artículo 7o del Decreto 843 de 2016 y en desarrollo del Decreto 677 de 1995, y
CONSIDERANDO:
Que el inciso segundo del artículo 245 de la Ley 100 de 1993 determina que el Gobierno nacional reglamentará, entre otros, el régimen de registros sanitarios de los productos objeto de competencia por parte del Instituto Nacional de Vigilancia de Medicamentos y Alimentos (Invima), dentro de los cuales se encuentran los medicamentos.
Que los requisitos para la obtención de los registros sanitarios de medicamentos de síntesis química están contemplados en el Decreto 677 de 1995 y sus modificatorias, norma que dispone en los artículos 22 literal n) y 31, numeral 1 y su parágrafo cuarto, que el interesado en obtener ante el Invima el correspondiente registro sanitario, deberá aportar los estudios de estabilidad que determinan el periodo de vida útil del producto.
Que mediante Resolución 2514 de 1995, el entonces Ministerio de Salud adoptó la guía práctica de requisitos para el desarrollo de estudios de estabilidad de medicamentos.
Que Colombia fue clasificada por la Organización Mundial de la Salud (OMS) en el informe 43 (“WHO Technical Report Series, número 953, 2009), en la zona climática IVb, por lo cual el Ministerio de Salud y Protección Social, de acuerdo con lo dispuesto en el artículo 7o del Decreto 843 de 2016, debe establecer los requisitos y criterios a que habrán de sujetarse los estudios de estabilidad que deben presentar los interesados en la obtención de registro sanitario para los medicamentos de síntesis química y su vigencia.
Que en consonancia con lo anterior, los estudios de estabilidad que presenten los interesados en obtener el registro sanitario deben tomar en cuenta esta clasificación para evaluar la variación en el tiempo de la calidad de los Ingredientes Farmacéuticos Activos (IFAs) y de los Productos Farmacéuticos Terminados (PFTs), con el propósito de establecer la vida útil y las condiciones de almacenamiento recomendadas.
Que la Dirección de Medicamentos y Tecnologías en Salud de este Ministerio en conjunto con el Invima revisó fuentes nacionales e internacionales y estructuró la guía de estabilidad de los medicamentos de síntesis química de acuerdo con el desarrollo científico en el ámbito farmacéutico y la actual clasificación de zona climática para Colombia, acogiendo las recomendaciones del Comité de expertos sobre las especificaciones farmacéuticas de la Organización Mundial de la Salud (OMS) y de la Conferencia Internacional de Armonización sobre los estudios de estabilidad para los medicamentos de síntesis química.
Que este Ministerio mediante Oficio número 201724001743561 y MinCIT número 1-2017-016131, solicitó concepto previo al Ministerio de Comercio, Industria y Turismo, en cumplimiento a lo dispuesto en el artículo 2.2.1.7.5.6. del Decreto 1074 de 2015, Único Reglamentario del Sector Comercio, Industria y Turismo, ante lo cual la Dirección de Regulación de dicho Ministerio, mediante radicado 2-2017-018462 recibido por este Ministerio el 22 de septiembre del 2017 con radicado 201742302104842, manifestó que “(…) a la luz del Acuerdo sobre Obstáculos Técnicos al Comercio de la Organización Mundial del Comercio, no tiene que ver con un reglamento técnico de producto, por ende no está sujeto a lo señalado en el artículo 2.2.1.7.5.6 del Decreto 1595 de 5 de agosto del 2015”.
En mérito de lo expuesto,
RESUELVE:
ARTÍCULO 1o. OBJETO. La presente resolución tiene por objeto expedir la “Guía para el desarrollo y presentación de los estudios de estabilidad de medicamentos de síntesis química” contenida en el anexo técnico que hace parte integral de la presente resolución.
ARTÍCULO 2o. ÁMBITO DE APLICACIÓN. La presente resolución aplica a:
2.1. Los interesados en obtener registro sanitario de medicamentos de síntesis química.
2.2. Los titulares de registro sanitario interesados en renovar los registros sanitarios de medicamentos de síntesis química.
2.3. Los titulares de registro sanitario, cuando se presenten modificaciones al mismo, por cambios en aspectos que tienen efecto sobre la estabilidad del medicamento.
2.4. Los titulares de registro sanitario, fabricantes e importadores de medicamentos de síntesis química.
PARÁGRAFO. Los destinatarios de la presente resolución garantizarán la estabilidad del medicamento de síntesis química en todo el proceso productivo, desde el ingrediente farmacéutico activo hasta el final de la vida útil del producto farmacéutico terminado, así como durante la vigencia del registro sanitario, en los términos del anexo que hace parte integral del presente acto administrativo.
ARTÍCULO 3o. RESPONSABILIDAD. <Artículo modificado por el artículo 1 de la Resolución 1839 de 2020. El nuevo texto es el siguiente:> Serán responsables del cumplimiento de las disposiciones aquí previstas, los titulares de los registros sanitarios, fabricantes e importadores de los medicamentos de síntesis química, que presenten los estudios de estabilidad ante el Invima, garantizando con ello la veracidad de la información que allí suministren y cumpliendo los requisitos señalados en esta resolución, incluyendo el anexo técnico que hace parte de este acto.
Para tal fin realizarán las adecuaciones técnicas e inversiones necesarias para garantizar su cumplimiento. El Invima brindará la asistencia que se requiera, hará seguimiento y podrá solicitar informes sobre el grado de implementación.
ARTÍCULO 4o. RESPONSABILIDAD EN LAS CONDICIONES DE ALMACENAMIENTO. Durante todo el tiempo de vida útil del medicamento, el titular de registro, el fabricante, el importador, el acondicionador, el envasador, el semielaborador, el distribuidor o cualquiera que intervenga en la cadena de fabricación, comercialización, distribución y utilización, deberá garantizar en lo que le compete, las condiciones de almacenamiento del producto aprobadas por el Invima.
ARTÍCULO 5o. ESTABILIDAD DEL INGREDIENTE FARMACÉUTICO ACTIVO (IFA). Los interesados en obtener el registro sanitario, su renovación o modificación no son responsables de la realización de los estudios de estabilidad del IFA, pero deben presentar la información de estabilidad del IFA contenida en el numeral 7.1, proveniente del fabricante del IFA.
Igualmente, deben cumplir las condiciones de almacenamiento y material de envase, definidas por el fabricante del IFA. En su defecto, los interesados pueden realizar estudios propios de estabilidad del IFA, en cuyo caso deben tener en cuenta las disposiciones contenidas en el numeral 2 del anexo técnico.
ARTÍCULO 6o. ESTABILIDAD PARA PRODUCTOS FITOTERAPÉUTICOS Y GASES MEDICINALES. La “Guía para el Desarrollo y Presentación de los Estudios de Estabilidad de Medicamentos”, adoptada en la Resolución 2514 de 1995, aplicará únicamente para gases medicinales y productos fitoterapéuticos.
ARTÍCULO 7o. VIGENCIA. <Artículo modificado por el artículo 2 de la Resolución 1839 de 2020. El nuevo texto es el siguiente:> El presente acto administrativo rige a partir de la fecha de su publicación y surte efectos a partir del 1 de abril del año 2024, para productos farmacéuticos terminados (PFT) e Ingredientes Farmacéuticos Activos (IFA).
Publíquese y cúmplase.
Dada en Bogotá, D. C., a 26 de julio de 2018.
El Ministro de Salud y Protección Social,
Alejandro Gaviria Uribe.
GUÍA PARA EL DESARROLLO Y PRESENTACIÓN DE LOS ESTUDIOS DE ESTABILIDAD DE MEDICAMENTOS DE SÍNTESIS QUÍMICA.
CONTENIDO
1. INTRODUCCIÓN
1.1. OBJETIVO DE ESTA GUÍA
1.2. ALCANCE DE LA GUÍA
1.3. PRINCIPIOS GENERALES
1.4. ESTUDIOS DE ESTABILIDAD ACEPTADOS EN COLOMBIA
2. ESTABILIDAD PARA INGREDIENTES FARMACÉUTICOS ACTIVOS (IFAS)
2.1. ASPECTOS GENERALES
2.2. ENSAYOS BAJO ESTRÉS
2.3. SELECCIÓN DE LOS LOTES
2.4. SISTEMA DE ENVASE Y CIERRE
2.5. ESPECIFICACIONES
2.6. FRECUENCIA DE ANÁLISIS
2.7. CONDICIONES DE ALMACENAMIENTO PARA EL ESTUDIO DE ESTABILIDAD
2.7.1. Caso general
2.7.2. IFAs destinados a ser almacenados en un refrigerador
2.7.3. IFAs destinados a ser almacenados en un congelador
2.7.4. IFAs destinados a ser almacenados por debajo de -20 °C
2.8. GARANTÍA DE LA ESTABILIDAD
2.9. EVALUACIÓN
2.10. LEYENDAS Y ETIQUETADO
2.11. ESTUDIOS DE ASEGURAMIENTO DE LA ESTABILIDAD (ON-GOING STABILITY)
3. ESTABILIDAD EN LOS PRODUCTOS FARMACÉUTICOS TERMINADOS (PFTS)
3.1. ASPECTOS GENERALES
3.2. OBJETIVOS DEL ESTUDIO DE ESTABILIDAD
3.3. FOTOESTABILIDAD
3.4. SELECCIÓN DE LOTES
3.5. ENVASE PRIMARIO Y CIERRE DEL PRODUCTO
3.6. ESPECIFICACIONES DEL PRODUCTO FARMACÉUTICO TERMINADO
3.7. FRECUENCIA DE LOS ENSAYOS
3.8. CONDICIONES DE ALMACENAMIENTO PARA EL ESTUDIO DE ESTABILIDAD
3.8.1. Caso general
3.8.2. PFTs envasados en recipientes impermeables
3.8.3. PFTs envasados en recipientes semipermeables
3.8.4. PFTs destinados a ser almacenados en un refrigerador
3.8.5. PFTs destinados a ser almacenados en un congelador
3.8.6. PFTs destinados a ser almacenados por debajo de -20 °C
3.8.7. Consideraciones adicionales para las condiciones de almacenamiento
3.9. GARANTÍA DE LA ESTABILIDAD
3.10. EVALUACIÓN
3.11. LEYENDAS Y ETIQUETADO
3.12. ESTABILIDAD EN USO
3.13. ESTUDIOS DE ESTABILIDAD EN LA ETAPA DE POSMERCADEO POR CAMBIOS QUE AFECTAN LA ESTABILIDAD
3.14. ESTUDIOS DE ASEGURAMIENTO DE LA ESTABILIDAD (ON-GOING STABILITY)
4. CONSIDERACIONES SOBRE EL DISEÑO DEL ESTUDIO DE ESTABILIDAD
4.1. ASPECTOS GENERALES
4.1.1. Metodología analítica indicadora de estabilidad
4.1.2. Temperaturas de almacenamiento
4.1.3. Calidad microbiológica
4.1.4. Productos de descomposición o degradación
4.1.5. Consideraciones sobre los ensayos a efectuar según la forma farmacéutica en estudio
4.1.6. Consideraciones de tipo estadístico
4.2. ASPECTOS ESPECÍFICOS
4.2.1. Consideraciones para el diseño experimental
4.2.2. Consideraciones a aplicar para el muestreo
4.2.3. Consideraciones sobre el número de muestras a utilizar
5. ANÁLISIS DE LOS DATOS E INTERPRETACIÓN DE LOS ESTUDIOS DE ESTABILIDAD (9)
6. ESTUDIOS DE ESTABILIDAD ESPECIALES
6.1. PARA APOYAR EXCURSIONES DE LAS CONDICIONES DE ALMACENAMIENTO Y DISTRIBUCIÓN
6.2. PRODUCTO A GRANEL, EN PROCESO O INTERMEDIO
7. PRESENTACIÓN DEL PROTOCOLO E INFORME DEL ESTUDIO DE ESTABILIDAD
7.1. ELEMENTOS DEL INFORME DEL ESTUDIO DE ESTABILIDAD DEL IFA
7.2. ELEMENTOS DE UN PROTOCOLO DE ESTUDIOS DE ESTABILIDAD DEL PFT
7.3. ELEMENTOS DEL INFORME DEL ESTUDIO DE ESTABILIDAD DEL PFT
8. GLOSARIO
9. APÉNDICE 1. ÁRBOL DE DECISIÓN
10. BIBLIOGRAFÍA
1. INTRODUCCIÓN
El presente documento se estructura tomando como referencia el Anexo 2 del Informe 43 de la OMS de 2009, las guías ICH sobre los requerimientos para el desarrollo y presentación de estudios de estabilidad y la Guía para el desarrollo y presentación de los estudios de estabilidad de medicamentos acogida mediante Resolución 2514 de 1995, desde el trámite del Registro Sanitario, hasta la verificación del cumplimiento de las BPM.
Los estudios de estabilidad para los medicamentos de síntesis química que se comercialicen en el territorio nacional deben cumplir con la temperatura de 30°C ± 2°C, y la humedad relativa de 75% HR ± 5% HR, de acuerdo con la Zona Climática IVb establecida por la Organización Mundial de la Salud (OMS), para Colombia.
1.1. OBJETIVO DE ESTA GUÍA
Este documento se constituye en un lineamiento para la realización y presentación de los estudios de estabilidad de los medicamentos de síntesis química.
1.2. ALCANCE DE LA GUÍA
Esta directriz se aplica a los ingredientes farmacéuticos activos (IFAs) y productos farmacéuticos terminados (PFTs) nuevos y a los ya comercializados de síntesis química, en cualquier modalidad de comercialización en el país. Por lo tanto, lo aquí descrito excluye los estudios de estabilidad de productos cosméticos, productos fitoterapéuticos, medicamentos homeopáticos, gases medicinales, radiofármacos, suplementos dietarios y productos biológicos.
1.3. PRINCIPIOS GENERALES
El propósito de un estudio de estabilidad es suministrar la evidencia de la variación en la calidad de un IFA o de un PFT en el tiempo por la influencia de diferentes factores ambientales, tales como la temperatura, la humedad y la luz, entre otros (1, 2). El programa de estudios de estabilidad también incluye la evaluación de los factores relacionados con el producto que influyen en su calidad, como, por ejemplo, la interacción del IFA con los excipientes, con el sistema de envase y cierre y los materiales de empaque, así como la incompatibilidad entre IFAs y entre excipientes dentro de una misma formulación, como por ejemplo entre las vitaminas de un producto multivitamínico, o entre preservantes o viscosantes (1).
Como resultado de estos estudios de estabilidad, se logra establecer el periodo de reanálisis y la vida útil de un IFA, la vida útil de un PFT y las condiciones de almacenamiento recomendadas según la zona de comercialización del medicamento (1,2). Adicional a esto, los estudios de estabilidad son el soporte fundamental para el desarrollo de un producto, así como para la vigilancia de su calidad en la etapa de posmercadeo.
1.4. ESTUDIOS DE ESTABILIDAD ACEPTADOS EN COLOMBIA
En Colombia se aceptan los siguientes tipos de estudios de estabilidad para presentar la solicitud, modificación o renovación de un registro sanitario de un medicamento, a saber:
a) Estudios de Estabilidad a largo plazo o natural
b) Estudios de Estabilidad a corto plazo o acelerados
c) Estudios de estabilidad en la etapa de posmercadeo
d) Estudios de aseguramiento de estabilidad (on-goingstability)
Los estudios de corto plazo (estabilidad acelerada) sirven para predecir la vida útil más probable (tentativa) del PFT en condiciones normales de almacenamiento y los estudios a largo plazo (estabilidad natural) sirven para establecer la vida útil definitiva del PFT bajo condiciones de almacenamiento definidas. Por esta razón, los primeros, los acelerados, se consideran provisionales y no son obligatorios si se cuenta con los estudios de largo plazo terminados hasta el final de la vida útil solicitada. Los estudios de estabilidad natural se consideran definitivos y son de carácter obligatorio.
2. ESTABILIDAD PARA INGREDIENTES FARMACÉUTICOS ACTIVOS (IFAs)
2.1. ASPECTOS GENERALES
La información sobre la estabilidad de un IFA es una parte integral del acercamiento sistemático para la evaluación de la estabilidad (1,2). Los atributos generales que deben ser evaluados en un IFA en un estudio de estabilidad son la apariencia, la cuantificación o la potencia y la concentración de los productos de descomposición. Sin embargo, se pueden utilizar otros parámetros susceptibles de alteración, dependiendo de las características del IFA, como puede ser el ensayo microbiológico (1).
El periodo de reanálisis de un IFA o la vida útil asignada por el fabricante se deriva necesariamente de los datos provenientes del estudio de estabilidad (1), por lo que no se puede establecer una vigencia o reanálisis del IFA más allá del citado por el fabricante del IFA, sin el respectivo soporte del estudio de estabilidad del mismo.
Los IFAs deben ser almacenados bajo las condiciones recomendadas y en el material de envase original, de acuerdo con los estudios de estabilidad realizados por el fabricante del mismo. De mantenerse estas condiciones en el lugar de almacenamiento del IFA, el Titular del PFT no está obligado a realizar los estudios de estabilidad del IFA, pero sí debe presentar los estudios de estabilidad realizados por el fabricante del IFA dentro de la documentación requerida para la obtención y renovación del registro sanitario o modificación del mismo por cambios que afectan la estabilidad y la información presentada debe contener como mínimo lo descrito en el numeral 7.1.
En caso de que el IFA no se almacene bajo las condiciones recomendadas por el fabricante del mismo o no se conserve en el material de envase original, dentro de la documentación requerida para la obtención y renovación del registro sanitario o modificación del mismo por cambios que afectan la estabilidad, se deben desarrollar y presentar los respectivos estudios de estabilidad a las condiciones de almacenamiento determinadas por la condición climática bajo la cual se destina almacenar el IFA hasta el fin de la vida útil establecida, teniendo en cuenta lo dispuesto en el numeral 2 y la información presentada debe contener como mínimo lo descrito en el numeral 7.1. Estos estudios suministran la información experimental del efecto del cambio de la condición de almacenamiento utilizada, sobre la estabilidad y calidad del IFA, de tal manera que los resultados obtenidos permitan tomar las decisiones correspondientes sobre la vida útil del IFA, así como su uso e impacto en el PFT.
2.2. ENSAYOS BAJO ESTRÉS
Los ensayos bajo estrés realizados en un IFA pueden ayudar a identificar los productos de degradación, lo que a su vez apoya en el establecimiento de las rutas de descomposición, la estabilidad intrínseca de la molécula y para validar la capacidad indicadora de estabilidad que posee el procedimiento analítico que se está diseñando para aplicar en los estudios de estabilidad. La naturaleza de los ensayos bajo estrés depende del IFA en particular y del tipo de PFT involucrado (1,2).
Se pueden utilizar los siguientes enfoques para la evaluación de un IFA (1):
a) Cuando exista información disponible, se acepta suministrar los datos importantes que se hayan publicado en la literatura científica con el fin de identificar los productos de descomposición, así como los mecanismos de reacción.
b) Cuando no haya información disponible, se deben desarrollar los estudios bajo estrés.
Los estudios bajo estrés se pueden llevar a cabo sobre un solo lote del IFA. Estos deben incluir el efecto de la temperatura, trabajando con incrementos de 10 °C (por ej. 50 °C, 60 °C, etc.) por encima de la temperatura utilizada para los ensayos acelerados (40 °C), la humedad (por ej. 75% de humedad relativa (HR) o mayor) y según el caso, la oxidación y la fotólisis del IFA. Los ensayos también deben evaluar la susceptibilidad del IFA a la hidrólisis cuando está en solución o suspensión, por medio de un estudio dentro de un rango de valores de pH justificados. La fotoestabilidad debe ser desarrollada como parte integral de los ensayos bajo estrés (1,2). Para la realización de estudios de fotoestabilidad ver la Guía ICH Q1B sobre "Ensayos de fotoestabilidad de nuevos IFAs y PFTs" (3).
Los resultados de estos ensayos forman parte integral de la información que se debe presentar a la autoridad regulatoria (1,2).
2.3. SELECCIÓN DE LOS LOTES
Se debe suministrar datos de los estudios de estabilidad provenientes de por lo menos tres lotes primarios fabricados del IFA. Los lotes deben ser fabricados por lo menos a la escala piloto industrial, empleando la misma ruta sintética que se sigue en los lotes de producción industrial y utilizando un método de manufactura y procedimiento que simule todo el proceso final que se emplea para la manufactura de lotes industriales (1,2).
La calidad global de los lotes de IFA colocados en estudio de estabilidad, debe ser representativa de la calidad del material que va a ser elaborado en escala de producción industrial (1,2). Para IFAs que se conoce científicamente que son estables, se puede presentar información proveniente de por lo menos dos lotes primarios (1).
2.4. SISTEMA DE ENVASE Y CIERRE
Los estudios de estabilidad para un IFA se deben conducir envasándolo en el mismo sistema de envase y cierre que se propone para su almacenamiento y distribución, o en uno que lo simule al máximo (1,2).
2.5. ESPECIFICACIONES
Los estudios de estabilidad deben incluir la evaluación de aquellos atributos del IFA que son susceptibles de cambiar durante el almacenamiento y que se conoce influyen sobre la calidad, seguridad y/o eficacia (1,2). El ensayo debe cubrir en forma apropiada los atributos físicos, químicos, biológicos (p.ej. potencia en antibióticos) y microbiológicos que fueron mencionados en el numeral 2.1.
Se debe utilizar un método analítico validado, específico e indicador de estabilidad. Los aspectos a replicar y la cantidad de réplicas que deben efectuarse dependerán de los resultados obtenidos en el estudio de validación (1,2).
Cabe resaltar que, dentro de las BPM, el fabricante de un PFT, es el responsable de verificar las especificaciones de calidad del IFA.
2.6. FRECUENCIA DE ANÁLISIS
Para los estudios de largo plazo, la frecuencia de análisis debe ser lo suficiente para cubrir el periodo de reanálisis o de vida útil propuesto para el IFA (1,2).
Para IFAs a los que se propone un periodo de reanálisis o una vida útil de por lo menos doce meses, la frecuencia de los ensayos en condiciones de almacenamiento de largo plazo, por lo general, será cada tres meses durante el primer año, cada seis meses en el segundo año y anualmente de ahí en adelante hasta llegar al periodo de reanálisis o vida útil propuesto (1,2).
A condiciones de almacenamiento aceleradas, se recomienda un estudio de seis meses que incluya por lo menos tres puntos de ensayo teniendo en cuenta el punto final y el inicial (p. ej., 0, 3 y 6 meses). Cuando se sospeche (con base en la experiencia adquirida) que los resultados de un estudio acelerado se aproximan al criterio de "cambio significativo", se debe incrementar el número de ensayos, bien sea adicionando muestras en el punto final o incluyendo un cuarto punto en el diseño del estudio (1,2).
Cuando la condición de almacenamiento intermedia se emplea como resultado de un cambio significativo en el estudio de estabilidad acelerado, se recomienda evaluar por lo menos cuatro puntos, que incluyan el inicial y el final (p. ej. 0, 6, 9, y 12 meses) para un estudio de doce meses (1,2).
2.7. CONDICIONES DE ALMACENAMIENTO PARA EL ESTUDIO DE ESTABILIDAD
Por lo general un IFA debe ser evaluado en las condiciones de almacenamiento (con tolerancias apropiadas), lo que permite evaluar su estabilidad térmica y si es aplicable, su sensibilidad a la humedad. Las condiciones de almacenamiento y la longitud de los estudios deben ser suficientes para cubrir el almacenamiento, la distribución y el uso (1,2).
Las tolerancias de las condiciones de almacenamiento se definen, como las variaciones aceptables en temperatura y humedad relativa de los sitios de almacenamiento para los estudios de estabilidad. Los equipos utilizados deben ser capaces de controlar las condiciones de almacenamiento, dentro de los rangos definidos en esta guía. Las condiciones de almacenamiento deben ser monitoreadas y registradas. Se aceptan como inevitables, los cambios ambientales cortos debidos a la apertura de las puertas del sitio de almacenamiento.
Se debe evaluar el efecto de las excursiones (temperatura y humedad relativa), debido a la falla de los equipos, diligenciando un reporte, si se considera que esto afecta los resultados de estabilidad. Las excursiones que exceden por más de 24 horas las tolerancias definidas, se deben describir en el informe del estudio, así como la evaluación de los efectos (1).
En el momento de la solicitud del Registro Sanitario del PFT, los estudios de estabilidad de largo plazo del IFA se deben haber desarrollado durante un mínimo de doce meses para el número de lotes especificados en el numeral 2.3 y los estudios se deben continuar por un tiempo suficiente para cubrir el periodo de reanálisis o de vida útil propuesto. Para los compuestos que se reconocen científicamente como estables, los datos mencionados deben cubrir un mínimo de seis meses en el momento de la solicitud de registro. Los datos adicionales acumulados durante el periodo de evaluación de la solicitud de registro, se deben entregar a la autoridad cuando lo requiera. Los datos provenientes de los estudios a condiciones aceleradas y si es apropiado, los provenientes de almacenamiento a condiciones intermedias, pueden ser utilizados para evaluar el efecto de las salidas accidentales cortas (excursiones) de las condiciones de almacenamiento etiquetadas (tal como puede ocurrir durante la distribución) (1,2).
Las condiciones de almacenamiento para los IFAs en los estudios a largo plazo, acelerados y cuando sea apropiado, intermedios, se detallan en los numerales 2.7.1 a 2.7.4. El llamado caso general aplica si el IFA no está específicamente cubierto por las condiciones indicadas en los numerales 2.7.2 a 2.7.4. (1,2).
2.7.1. Caso general
Tabla 1. Caso general (1,2)

* El que los estudios de estabilidad naturales se realicen a 25°C ± 2°C/60% HR ± 5% HR o 30°C ± 2°C/65% HR ± 5% HR o 30°C ± 2°C/75% HR ± 5% HR, está determinado por la condición climática bajo la cual se destina almacenar el IFA. Un ensayo a una condición de estabilidad de largo plazo más severa puede ser una alternativa para el almacenamiento a 25°C ± 2°C/60% HR ± 5% HR o 30°C ± 2°C/65% HR ± 5% HR.
** Si la condición del estudio a largo plazo es 30°C ± 2°C/65% HR ± 5% HR, o 30°C ± 2°/75%HR, no existe una condición intermedia.
Si los estudios de largo plazo, por ejemplo, son desarrollados a 25°C ± 2°C/60%HR ± 5%HR y ocurre un "cambio significativo" en cualquier momento durante el estudio de seis meses a condiciones aceleradas, se debe realizar un ensayo adicional a la condición de almacenamiento intermedia y evaluarla en relación con el criterio de cambio significativo. En este caso, los estudios a condiciones de almacenamiento intermedio deben incluir todos los ensayos que se efectúan en los de largo plazo, a menos que haya otra justificación y se debe incluir, como mínimo, datos de seis meses provenientes de un estudio proyectado a 12 meses, realizado a condiciones intermedias de almacenamiento (1,2).
Para un IFA, "cambio significativo" se define como una falla en el cumplimiento de sus especificaciones (1,2).
2.7.2. IFAs destinados a ser almacenados en un refrigerador
Tabla 2. IFAs destinados para almacenamiento en el refrigerador (1,2).
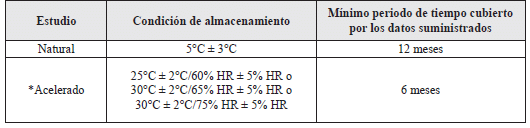
* El que los estudios de estabilidad acelerados se realicen a 25°C ± 2°C/60% HR ± 5% HR o 30°C ± 2°C/65% HR ± 5% HR o 30°C ± 2°C/75% HR ± 5% HR, está fundamentado en la evaluación del riesgo. Un ensayo a una condición de estabilidad de largo plazo más severa puede ser una alternativa para el almacenamiento a 25°C ± 2°C/60% HR ± 5% HR o 30°C ± 2°C/65% HR ± 5% HR.
Los datos bajo refrigeración se deben evaluar de acuerdo con lo indicado en el numeral 2.9 de evaluación de esta guía, excepto donde explícitamente aplica lo que se menciona a continuación:
a) Si ocurre un cambio significativo entre los tres y los seis meses del ensayo a condiciones de almacenamiento acelerado, el periodo de reanálisis que se proponga, debe estar fundamentado en los datos disponibles en condiciones de almacenamiento de largo plazo (1,2).
b) Si ocurre el cambio significativo, dentro de los tres primeros meses del ensayo a la condición del almacenamiento acelerado, se debe suministrar un análisis crítico (argumentación) de la situación, para orientar sobre el efecto que presentaría la salida accidental por corto tiempo, por fuera de las condiciones de almacenamiento etiquetadas, por ejemplo, durante la distribución o el almacenamiento. Esta argumentación puede ser sustentada si se dispone de datos apropiados, por un periodo menor de tres meses, provenientes de ensayos adicionales realizados sobre un solo lote del IFA, pero con una mayor frecuencia de análisis de la usual. Cuando ha ocurrido un cambio significativo dentro de los tres primeros meses, se considera innecesario continuar con el ensayo del IFA hasta completar los seis meses (1,2).
2.7.3. IFAs destinados a ser almacenados en un congelador
Tabla 3. IFAs destinados para almacenamiento en el congelador (1,2).

En el caso extraordinario de que un IFA, sea destinado para almacenar en congelación, el periodo de reanálisis o la vida útil, debe estar basada en los datos obtenidos en las condiciones de almacenamiento de largo plazo. En ausencia de un almacenamiento en condiciones aceleradas para el IFA, destinado a ser almacenado en un congelador, se debe realizar un ensayo sobre un solo lote a una temperatura elevada (por ejemplo 5°C ± 3ºC o 25ºC ± 2ºC o 30ºC ±2 ºC) por un periodo de duración apropiado, para orientar sobre el efecto de las salidas de corto plazo por fuera de las condiciones de almacenamiento propuestas, por ejemplo, durante la distribución o el almacenamiento (1,2).
2.7.4. IFAs destinados a ser almacenados por debajo de -20 ºC
Deben ser manejados sobre la base de estudio del caso en particular (1,2).
2.8. GARANTÍA DE LA ESTABILIDAD
Cuando los datos del estudio de estabilidad de largo plazo realizados sobre lotes primarios del IFA no cubran el periodo de reanálisis o vida útil propuesto, asegurado en el momento de la solicitud, se deben presentar los respectivos soportes que garanticen la continuación y finalización de los estudios de estabilidad con posterioridad a la aprobación de la solicitud, con el fin de establecer, en forma segura, el periodo de reanálisis o la vida útil del IFA en cuestión (1,2). La documentación correspondiente al proceso se debe entregar cuando se culmine y cuando la autoridad lo requiera.
Cuando la solicitud incluya datos del estudio de estabilidad de largo plazo sobre el número de lotes de producción especificado en el numeral 2.3 y cubren el periodo de reanálisis o vida útil propuesto, no se considera necesario tomar una acción de garantía de la estabilidad. De lo contrario, se puede tomar una de las siguientes acciones (1,2):
a) Si la solicitud incluye datos provenientes del estudio de estabilidad sobre el número de lotes de producción especificado en el numeral 2.3, los cuales no han finalizado, se debe continuar estos estudios a lo largo del periodo de reanálisis o vida útil propuesta.
b) Si la solicitud incluye datos provenientes del estudio de estabilidad realizado sobre un número de lotes de producción inferior al especificado en el numeral 2.3, se debe continuar con estos estudios a lo largo del periodo de reanálisis o vida útil propuesto y colocar lotes de producción adicionales, hasta un total de por lo menos tres, en estudios de estabilidad de largo plazo hasta cubrir el periodo de reanálisis o vida útil propuesto.
c) Si la solicitud no incluye datos de estabilidad sobre lotes de producción, se debe iniciar el estudio de estabilidad de largo plazo con dos o tres lotes de producción (ver el numeral 2.3) y se debe desarrollar hasta el periodo de reanálisis o vida útil propuesto.
El protocolo de estabilidad utilizado para los estudios de largo plazo, dentro de las actividades de garantía de estabilidad, debe ser el mismo que se estableció para los lotes primarios, a menos que científicamente se justifique otra cosa (1,2).
2.9. EVALUACIÓN
El propósito del estudio de estabilidad del IFA es establecer un periodo de reanálisis o vida útil aplicable a todos los lotes futuros del IFA, fabricados bajo circunstancias similares, con base en los ensayos de un mínimo número de lotes del IFA según lo especificado en la sección 2.3 (a menos que se justifique y se conceda un número de lotes diferente por parte de la autoridad sanitaria competente) y la evaluación de la información de estabilidad donde se incluya, según corresponda, resultados de los ensayos físicos, químicos, biológicos y microbiológicos. El grado de variabilidad de los lotes individuales, afecta la confiabilidad de que la producción futura de lotes permanecerá dentro de las especificaciones a lo largo del periodo de reanálisis o vida útil asignado (1,2).
Los datos pueden presentar tan poca degradación y variabilidad, que aparentemente con solo observarlos se podría garantizar el periodo de reanálisis o vida útil solicitada. Bajo esta circunstancia, por lo general es innecesario someter la información a un análisis estadístico; sería suficiente para omitir este tratamiento, el suministrar una justificación técnica apropiada (1,2).
Una aproximación para el análisis de los datos de un atributo cuantitativo que se espera cambie con el tiempo, es determinar el momento en el que el límite de confianza del 95% de una sola cola para la curva promedio, intercepta el criterio de aceptación. Si el análisis muestra que la variabilidad lote a lote es pequeña, es ventajoso combinar los datos y hacer una sola estimación global. Esto se puede hacer si se aplica primero un ensayo estadístico apropiado (p. ej. valores de p para un nivel de significancia de rechazo de más de 0,25) a las pendientes de las líneas de regresión y a los interceptos a tiempo cero para los lotes individuales. Si no es apropiado combinar los datos de lotes diferentes, el periodo de reanálisis o vida útil global debería basarse en el tiempo mínimo al que un lote se esperaría pudiera permanecer dentro de los criterios de aceptación (criterio del peor caso) (1,2).
La naturaleza de cualquier relación de descomposición, determinará cuándo los datos deben ser transformados para un análisis de regresión lineal. Usualmente las relaciones pueden ser representadas por una función rectilínea, cuadrática o cúbica sobre una escala aritmética o logarítmica. Tanto como sea posible, la selección de un modelo debería justificarse por un razonamiento físico y/o químico y también tendría en cuenta la cantidad de datos disponibles (principio de parsimonia), para asegurar una predicción robusta. Los métodos estadísticos deben ser empleados para evaluar la bondad de ajuste de los datos de todos los lotes y los lotes combinados (donde sea apropiado), a la línea o curva de descomposición supuesta (1,2).
Si se justifica, se puede extrapolar el límite de los datos provenientes del estudio en condiciones de largo plazo, más allá del rango de observación, para extender el periodo de reanálisis o vida útil. Esta justificación debería estar fundamentada en lo que se conoce acerca del mecanismo de degradación, los resultados del ensayo bajo condiciones aceleradas, la bondad de ajuste a cualquier modelo matemático, el tamaño de lote y la existencia de datos de estabilidad de soporte. Sin embargo, esta extrapolación supone que la misma relación de descomposición continuará aplicándose más allá de los datos observados (1,2).
Cualquier evaluación deberá cubrir no solamente la valoración, sino también los niveles de los productos de descomposición y otros atributos apropiados (1,2). Donde sea conveniente, se debe poner atención a la revisión de la adecuación de la evaluación que une la estabilidad del producto farmacéutico terminado y el comportamiento de degradación durante el estudio (1). Cuando existan restricciones sobre el contenido de un producto de descomposición, se debe tener en cuenta este criterio en conjunto con el límite de descomposición del IFA, y la vida útil o periodo de reanálisis estará determinado por el criterio que se cumpla primero.
2.10. LEYENDAS Y ETIQUETADO
Con base en la evaluación de la estabilidad del IFA, el fabricante del IFA debe establecer un informe que especifique las condiciones para el almacenamiento, con el fin de colocar la información pertinente en la etiqueta. Donde sea aplicable, se deben suministrar instrucciones específicas en particular para el IFA, por ejemplo, que no puede tolerar enfriamiento o salidas de las condiciones de temperatura de almacenamiento especificadas (1,2).
El periodo de reanálisis o vida útil se deriva de la información de estabilidad y se debe desplegar una fecha de reanálisis o vida útil sobre la etiqueta del envase (1,2).
2.11. ESTUDIOS DE ASEGURAMIENTO DE LA ESTABILIDAD (ON-GOING STABILITY)
La estabilidad del IFA debe ser verificada de acuerdo con un programa continuo y apropiado que permita la detección de cualquier evento de inestabilidad (p.ej. cambios en los niveles de los productos de descomposición). El propósito del programa de Aseguramiento de Estabilidad es monitorear el comportamiento del IFA y determinar que este permanece y puede permanecer dentro de las especificaciones bajo las condiciones de almacenamiento indicadas en la etiqueta, dentro del periodo de reanálisis o vida útil en todos los lotes futuros (1).
El programa de Aseguramiento de Estabilidad se debe presentar en un protocolo escrito y los resultados se mostrarán en un reporte formal (1).
El protocolo para un programa de Aseguramiento de Estabilidad se debe prolongar hasta el final del periodo de reanálisis o vida útil y debe incluir, pero no está limitado a los siguientes parámetros (1):
a) Número(s) lote(s) y diferentes tamaños de lotes si aplica
b) Métodos de ensayo físicos, químicos, microbiológicos y biológicos, relevantes
c) Criterios de aceptación
d) Referencias para los métodos de ensayo
e) Descripción del sistema(s) de envase y cierre
f) Frecuencia de muestreo
g) Descripción de las condiciones de almacenamiento (se deben utilizar condiciones estandarizadas para ensayos de largo plazo, como se describe en esta guía y consistentes con el etiquetado del IFA)
h) Otros parámetros aplicables especificados para el IFA.
Por lo menos un lote de producción por año del IFA (a menos que ninguno se produzca durante el año), debe ser adicionado al programa de monitoreo de estabilidad y ensayado por lo menos anualmente para confirmar la estabilidad. En ciertas situaciones se deben incluir lotes adicionales en el programa de estabilidad "on-going". Por ejemplo, un estudio de estabilidad "on-going" se debe desarrollar después de cualquier cambio significativo o desviación significativa de la ruta de síntesis, del proceso o del sistema de envase y cierre, el que puede tener un impacto sobre la estabilidad del IFA (1).
Se deben investigar los resultados que se salen de las especificaciones o tendencias atípicas significativas. Todo cambio significativo confirmado, resultados que se salen de especificación o tendencias atípicas significativas, se deben reportar inmediatamente al respectivo fabricante del PFT. El posible impacto sobre los lotes en el mercado se debe evaluar con los respectivos fabricantes de los productos terminados y la autoridad competente (1).
Se debe escribir y mantener un resumen de todos los datos generados dentro del programa, incluyendo cualquier conclusión intermedia. Este resumen debe estar sujeto a revisiones periódicas y estar disponibles para inspecciones en cualquier momento por la entidad regulatoria (1).
Las condiciones estándar específicas para lo mencionado de estabilidad del IFA, el análisis de datos e interpretación de los estudios de estabilidad y los ejemplos de enfoques estadísticos para el análisis de datos (descritos en la guía ICH Q1E) pueden ser consultadas en la página web del Invima en el apartado de estabilidad con sus respectivas actualizaciones.
3. ESTABILIDAD EN LOS PRODUCTOS FARMACÉUTICOS TERMINADOS (PFTs)
3.1. ASPECTOS GENERALES
El diseño de los estudios de estabilidad para el PFT se debe basar en el conocimiento del comportamiento y propiedades del IFA, información proveniente de los estudios de estabilidad del IFA y de la experiencia ganada durante los estudios de preformulación e investigación del PFT (1,2).
3.2. OBJETIVOS DEL ESTUDIO DE ESTABILIDAD
El análisis de los resultados de los estudios de estabilidad realizados sobre el PFT, permite:
a) La evaluación de la vida útil del PFT.
b) El establecimiento de las condiciones de almacenamiento del PFT y donde sea necesario (por ejemplo, productos para reconstituir antes del uso), las condiciones para antes y después de la apertura del envase.
c) Generar una justificación adicional de cualquier porcentaje de exceso del IFA, necesario para garantizar la potencia y/o concentración del mismo, durante la vida útil propuesta para el PFT.
3.3. FOTOESTABILIDAD
Las pruebas de fotoestabilidad deben realizarse en al menos un lote primario del PFT, si procede (1,2,3). Las condiciones estándar para las pruebas de fotoestabilidad son descritas en la guía ICH Q1B.
3.4. SELECCIÓN DE LOTES
Los datos provenientes de los estudios de estabilidad se deben suministrar sobre la base de por lo menos tres lotes primarios del PFT. Los lotes primarios deben tener la misma formulación y el mismo sistema de envase y cierre, como se propone para la comercialización. El proceso de manufactura utilizado para los lotes primarios, debe simular en todo al que va a ser aplicado para los lotes de producción industrial, garantizando que el PFT sea de la misma calidad y cumpla las mismas especificaciones que se indican para el producto de comercialización (1,2). En el caso de formas farmacéuticas con IFAs que se conoce por evidencia científica que son estables, se deben suministrar datos provenientes de la evaluación de por lo menos dos lotes primarios (1).
Si se justifica, dos de los tres lotes deben ser por lo menos lotes de escala piloto industrial y el tercero puede ser menor. Donde sea posible, los lotes del PFT deben ser fabricados utilizando diferentes lotes del IFA (1,2).
Los estudios de estabilidad se deben desarrollar sobre cada concentración individual, forma de dosificación y tipo y tamaño de envase del PFT, a menos que se pueda aplicar un diseño de extremos o matrices (1,2,4). Las condiciones estándar para aplicar un diseño de extremos o matrices son descritas en la guía ICH Q1D.
3.5. ENVASE PRIMARIO Y CIERRE DEL PRODUCTO
Los estudios de estabilidad deben ser desarrollados para cada uno de los tipos de material de envase primario y cierre propuestos para la comercialización del producto (cuando sea apropiado cualquier empaque secundario o etiqueta) (1,2), incluyendo los envases de seguridad diseñados para evitar riesgos a los niños, como los diferentes tipos de cierre desarrollados para impedir la violación de la integridad del producto a pesar de la similitud que pueda existir en las subtapas de los envases. Las muestras médicas también deben incluirse en los estudios de estabilidad si su envase primario y cierre es diferente al empleado para el producto comercial.
Se debe evaluar, cuando sea posible y/o necesaria, la interacción que puede existir entre el medicamento, su envase primario y su cierre y la incorporación de materiales extractables y lixiviables en la composición del producto durante el almacenamiento, por medio de procedimientos muy sensibles y cuantificables. Estas pruebas son necesarias aún si el envase primario y cierre cumplen los ensayos apropiados, como los indicados en las farmacopeas para envases plásticos y cierres de caucho o plástico (5,6).
Se deben presentar los datos de estabilidad correspondientes para todos los tamaños de envases de dosis múltiple, tales como parenterales, aerosoles, oftálmicos, orales, etc. Lo anterior, aplica siempre y cuando no se cumplan las condiciones para emplear los diseños de estudios de estabilidad de extremos o de matrices descritos en la Guía ICH Q1D. En los casos que aplique para los PFTs en las formas farmacéuticas sólidas, se deben presentar los datos de estabilidad para los envases primarios y cierres (más grande y más pequeño) que van a ser comercializados, siempre y cuando, cualquier otro envase primario y cierre de tamaño intermedio sea de composición idéntica a los anteriores.
3.6. ESPECIFICACIONES DEL PRODUCTO FARMACÉUTICO TERMINADO
Los estudios de estabilidad deben incluir los ensayos de aquellos atributos del PFT que son susceptibles de cambiar durante el almacenamiento y que por lo tanto pueden influir en la calidad, seguridad y/o eficacia. Los ensayos deben cubrir tanto como sea apropiado, los atributos físicos, químicos, biológicos (p.ej. potencia en antibióticos) y microbiológicos, contenido de preservantes (p. ej. antioxidantes o preservantes antimicrobianos) y ensayos de funcionalidad (p. ej. para un sistema de liberación de dosis en aerosol). Los procedimientos analíticos deben estar completamente validados (para metodologías propias) o verificados (en caso de ser farmacopeicos) y ser específicos e indicadores de estabilidad. Dependiendo de los resultados de los estudios de validación correspondientes, para cada ensayo se debe establecer dónde y en qué cantidad se requiere desarrollar réplicas por cada muestra (1,2).
Los criterios para aceptación de la vida útil se deben derivar de la consideración de toda la información de estabilidad disponible. Pueden existir diferencias justificables entre los criterios de aceptación para vida útil y de liberación, con base en los resultados de la evaluación de la estabilidad y los cambios observados durante el almacenamiento. Cualquier diferencia entre el criterio de liberación y de vida útil para el contenido de preservantes antimicrobianos, debe estar soportada por una correlación validada del contenido químico y de la efectividad preservadora. Esto debe ser demostrado durante el desarrollo del producto farmacéutico con la formulación (excepto para la concentración preservadora), propuesta para su comercialización (1,2).
Para determinar la efectividad del preservante antimicrobiano se debe evaluar un lote primario de estabilidad del PFT al inicio y al final de la vida útil propuesta (además del contenido de preservantes), con el propósito de verificarla, sin tener en cuenta si hay una diferencia entre el criterio de liberación y de vida útil para el contenido del preservante (1,2).
3.7. FRECUENCIA DE LOS ENSAYOS
Para los estudios de largo plazo, la frecuencia de los ensayos debe ser suficiente para establecer el perfil de estabilidad del PFT (1,2).
Para un PFT con un periodo de vida útil de por lo menos doce meses, la frecuencia de los ensayos en las condiciones de almacenamiento de largo plazo, por lo general, sería cada tres meses durante el primer año, cada seis meses durante el segundo año y anualmente, de ahí en adelante, hasta cubrir la vida útil que se propone (1,2).
A condiciones de almacenamiento aceleradas, se recomienda un estudio de seis meses con un mínimo de tres puntos de tiempo de muestreo teniendo en cuenta el punto final y el inicial (p. ej. 0, 3 y 6 meses). Cuando se sospeche (con base en la experiencia desarrollada) que los resultados de un estudio acelerado se aproximan al criterio de "cambio significativo", se debe incrementar el número de ensayos, bien sea adicionando muestras en el punto final o incluyendo un cuarto punto en el diseño del estudio (1,2).
Si se justifica apropiadamente, se pueden aplicar diseños estadísticos experimentales reducidos de extremos o matrices (ver guía ICH Q1D), donde la frecuencia de muestreos y ensayos se reduce o ciertas combinaciones de factores no se evalúan en todas las muestras (1,2,4).
3.8. CONDICIONES DE ALMACENAMIENTO PARA EL ESTUDIO DE ESTABILIDAD
Por lo general, un PFT debe ser evaluado bajo condiciones de almacenamiento con tolerancias específicas que prueban su estabilidad térmica y si es aplicable, su sensibilidad a la humedad o a la pérdida potencial del solvente. Las condiciones de almacenamiento y la duración de los estudios escogidos, deben ser lo suficiente para cubrir el tiempo de almacenamiento, la distribución y el uso subsecuente, teniendo en cuenta las condiciones climáticas en las que el producto pretende ser comercializado (1,2).
Es necesario incluir en el protocolo la orientación del producto durante el almacenamiento, por ejemplo, de pie vs. invertido o acostado, en aquellos casos donde se espera que el contacto del producto con el sistema del cierre, pueda afectar la estabilidad del producto contenido o donde se haya hecho un cambio en el sistema de envase/cierre del producto (1).
Las condiciones de almacenamiento deben ser monitoreadas y registradas. El equipo utilizado debe ser capaz de controlar las condiciones de almacenamiento dentro de los rangos definidos en esta guía. Se aceptan y son inevitables los cambios ambientales de corta duración debido a la apertura de las puertas de las instalaciones de almacenamiento. Los efectos de las desviaciones causadas por las fallas del equipo se deben evaluar, analizar y reportar, si se juzga que afectan los resultados de estabilidad. Las desviaciones (excursiones) que exceden las tolerancias definidas por más de 24 horas se deben describir en el informe del estudio y se deben evaluar sus efectos (1).
Los ensayos de largo plazo deben cubrir por lo menos una duración de doce meses, en el momento de presentar la solicitud de registro sanitario y se deben continuar por un periodo de tiempo lo suficientemente largo para cubrir la vida útil propuesta (1,2). Para un PFT que contiene un IFA que se conoce científica y experimentalmente que es estable y donde no se observan cambios significativos en los estudios de estabilidad del PFT a condiciones aceleradas y a condiciones de largo plazo hasta los seis meses, se pueden presentar los datos que cubran por los menos seis meses de duración (1).
Los datos adicionales acumulados durante el periodo de evaluación de la solicitud del registro, se deben someter a evaluación de la autoridad sanitaria si lo requiere. Los datos provenientes de las condiciones aceleradas de almacenamiento se pueden utilizar para evaluar el efecto de las desviaciones de corta duración por fuera de las condiciones de almacenamiento etiquetadas (tal como puede ocurrir durante la distribución). Las condiciones de almacenamiento para el PFT, de largo plazo y aceleradas, se detallan en los numerales 3.8.1 a 3.8.6. El caso general se aplica si el PFT no está específicamente cubierto por las condiciones indicadas en los numerales 3.8.2 al 3.8.6. (1,2).
3.8.1. Caso general
Tabla 4. Caso general (1,2,)

Si en los estudios de estabilidad acelerada desarrollados ocurre un "cambio significativo", la estabilidad estaría determinada por los datos a largo plazo y la extrapolación no se considera apropiada. Por lo general se define cambio significativo para un PFT, en el estudio acelerado, como (1,2):
a) Un cambio del contenido inicial del IFA o IFAs del 5% o más, detectado en la valoración, o una falla para cumplir los criterios de aceptación para potencia, cuando se utilizan procedimientos biológicos o inmunológicos. (NOTA: Si se justifica se pueden aplicar otros valores para algunos productos, tales como multivitamínicos). El cambio significativo obedece a la mitad de degradación total esperada. Así, por ejemplo, si la especificación es 90-110%, se espera máximo una degradación del 10% y el cambio significativo es de 5% o más. Pero, si la especificación es 95-105%, se espera máximo una degradación del 5% y el cambio significativo es de 2,5% o más en la valoración.
b) Cualquier producto de descomposición que exceda su criterio de aceptación.
c) Fallas para el cumplimiento de los criterios de aceptación con relación a la apariencia, los atributos físicos y los ensayos de funcionalidad (p. ej.: color, separación de fases, redispersabilidad, compactación, dureza, dosis liberada por aplicación). Sin embargo, se pueden esperar algunos cambios en los atributos físicos (por ejemplo, ablandamiento de los supositorios diseñado para fundirse a 37 °C, si el punto de fusión está demostrado claramente, fusión de las cremas, pérdida parcial de la adhesión para un producto transdérmico), bajo condiciones aceleradas y en estas circunstancias, no se considera como un cambio significativo.
También se entiende como cambio significativo, cuando sea apropiado para la forma farmacéutica (1,2):
d) Fallas para cumplir el criterio de aceptación de pH.
e) Fallas para cumplir el criterio de aceptación de doce unidades en el ensayo de disolución.
En estudios de estabilidad natural, si en cualquier tiempo del estudio los resultados en el test de valoración o potencia están por fuera de las especificaciones establecidas o sucede lo descrito en los literales b, c, d y e del presente numeral, es un indicador de fallas en el PFT sometido a estudio, lo que requiere los respectivos ajustes (p.ej. formulación, diseño, envase, etc.) para someter nuevamente el PFT a evaluación.
3.8.2. PFTs envasados en recipientes impermeables
Los parámetros requeridos para clasificar los materiales de envase como permeables o impermeables, dependen de las características del material de envase, tales como el espesor y el coeficiente de permeabilidad. La adecuación del material de envase utilizado para un producto en particular, está determinada por las características del producto. Los envases considerados generalmente como resistentes a la humedad incluyen las ampollas de vidrio (1).
La sensibilidad a la humedad o a la pérdida potencial de solvente no es una preocupación del envase para PFTs en recipientes impermeables, que son los que suministran una barrera permanente al paso de la humedad o del solvente. Estos estudios de estabilidad para productos almacenados en envases impermeables pueden ser desarrollados bajo cualquier condición controlada o de humedad relativa ambiental (1,2).
3.8.3. PFTs envasados en recipientes semipermeables
Los productos fundamentalmente acuosos envasados en recipientes semipermeables, se deben evaluar para establecer la pérdida potencial de agua además de la estabilidad física, química, biológica y microbiológica. Esta evaluación puede ser llevada a cabo bajo condiciones de baja humedad relativa como se discute a continuación. Finalmente, se debe demostrar que los PFTs esencialmente acuosos almacenados en recipientes semipermeables, pueden resistir ambientes con baja humedad relativa. Se pueden desarrollar e informar otras aproximaciones comparables para productos no esencialmente acuosos (1,2).
Tabla 5. Condiciones para estudio de estabilidad en recipientes semipermeables para evaluar potenciales pérdidas de agua (1,2).

* (NMD = no más de… equivalente al NMT de la ICH).
Cuando los productos cumplen tanto las condiciones de largo plazo y las condiciones aceleradas, como se especificó en la tabla anterior, han demostrado la integridad del empaque en envases semipermeables. Un cambio significativo tan solo en la pérdida de humedad en las condiciones de almacenamiento acelerado, no necesita ser ensayado en otras condiciones de almacenamiento. Sin embargo, se deben suministrar datos para demostrar que el producto farmacéutico no tendría una pérdida significativa de agua a lo largo de la vida útil propuesta si es almacenado a 30°C/35% de HR (1).
Se considera que un cambio es significativo para un producto envasado en un recipiente semipermeable si la pérdida en agua es de un 5% con relación a su valor inicial, después de un almacenamiento equivalente de 3 meses a 40°C y no más del 25% de humedad relativa. Sin embargo, si se justifica, puede ser aceptable para los envases pequeños (un mililitro o menos) o productos unidosis, una pérdida de agua del 5% o más después de un almacenamiento equivalente de 3 meses a 40°C y no más del 25% de humedad relativa (1,2).
Una aproximación alternativa a los estudios desarrollados a humedad relativa baja, como se recomendó en la tabla anterior (tanto para ensayos de largo plazo como acelerados) es desarrollar los estudios de estabilidad bajo una humedad relativa mayor y relacionar la pérdida de agua por medio de cálculos a una humedad relativa menor. Esto puede lograrse mediante la determinación experimental del coeficiente de permeación para el sistema de envase cierre o, como se demuestra en el ejemplo siguiente, utilizando la relación calculada de las velocidades de pérdida de agua entre las dos condiciones de humedad a la misma temperatura. El coeficiente de permeación para un sistema de envase cierre puede ser determinado experimentalmente utilizando el escenario del peor caso, por ejemplo, evaluando la más diluida de una serie de concentraciones para el PFT propuesto (1,2).
3.8.3.1. Ejemplo de una aproximación para la pérdida de agua
Para un producto en un sistema de envase cierre determinado, tamaño de envase y llenado, una aproximación apropiada para obtener la velocidad de pérdida de agua a una condición de baja humedad relativa, es multiplicar la velocidad de pérdida de agua medida a una humedad relativa alternativa a la misma temperatura, por un coeficiente de velocidad de pérdida de agua mostrado en la tabla siguiente. Se debe demostrar que existe una velocidad de pérdida de agua lineal a la humedad relativa alternativa, durante el periodo de almacenamiento (1,2).
Tabla 6. Coeficientes para calcular la velocidad de pérdida de agua (1,2).
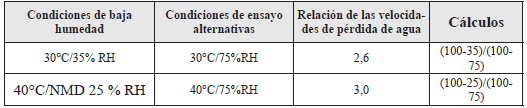
Por ejemplo, a una temperatura dada, sea el caso 40°C, la velocidad de pérdida de agua durante el almacenamiento a no más de 25% de humedad relativa, es la velocidad de pérdida de agua medida al 75% de humedad relativa multiplicada por tres, que es el correspondiente coeficiente de velocidad de pérdida de agua (1,2).
También se pueden utilizar coeficientes de velocidad de pérdida de agua válidos a condiciones de humedad relativa diferentes a aquellos que se muestran en la tabla anterior (1,2).
3.8.4. PFTs destinados a ser almacenados en un refrigerador
Se debe suministrar información apropiada para evaluar la cantidad de pérdida de agua, si el PFT está envasado en un recipiente semipermeable. Se deben evaluar los datos provenientes del almacenamiento bajo refrigeración de acuerdo con lo indicado en la sección de evaluación de esta guía, excepto donde explícitamente se indique lo que se describe a continuación (1,2).
Tabla 7. Condiciones para estudios de estabilidad en refrigerador (1,2).
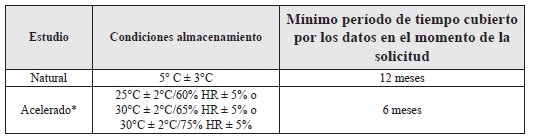
* El que los estudios acelerados se realicen a 25°C ± 2°C/60% HR ± 5% o 30°C ± 2°C/65% HR ± 5% o 30°C ± 2°C/75% HR ± 5%, está fundamentado en la evaluación basada en el riesgo. El ensayo a unas condiciones aceleradas más severas puede ser una alternativa para las condiciones de almacenamiento a 25°C/60% HR o 30°C/65% HR.
Si ocurre un cambio significativo entre el tercero y el sexto mes de un ensayo a condiciones de almacenamiento aceleradas, la vida útil propuesta debe estar fundamentada en los datos disponibles provenientes de un ensayo en condiciones de almacenamiento de largo plazo (1,2).
Si ocurre un cambio significativo dentro de los primeros tres meses de un ensayo en condiciones de almacenamiento aceleradas, se debe suministrar un análisis para explicar el efecto de las desviaciones de corta duración por fuera de las condiciones de almacenamiento etiquetadas, por ejemplo, durante la distribución y el almacenamiento. Si es apropiado, este análisis puede estar soportado por ensayos adicionales en un lote del PFT por un periodo menor a tres meses, pero con una frecuencia de muestreo mayor a la usual. Se considera que no es necesario continuar evaluando un producto hasta los seis meses, cuando ha ocurrido un cambio significativo dentro de los tres primeros meses del estudio acelerado, a la condición específica escogida, en concordancia con el análisis de riesgo (1,2).
3.8.5. PFTs destinados a ser almacenados en un congelador
Tabla 8. Condiciones para estudios de estabilidad en congelador (1,2).

Para PFTs destinados a ser almacenados en un congelador, la vida útil debe estar fundamentada en los datos obtenidos a largo plazo en las condiciones de almacenamiento del estudio natural. En ausencia de una condición de almacenamiento acelerada para PFTs destinados a ser almacenados en un congelador, se deben desarrollar ensayos sobre un solo lote a una temperatura elevada (sea el caso 5°C ± 3°C o 25°C ± 2°C o 30°C ± 2°C) por un periodo de tiempo apropiado, para poder orientar sobre el efecto de las excursiones de corto plazo por fuera de las condiciones de almacenamiento propuestas en la etiqueta (1,2).
3.8.6. PFTs destinados a ser almacenados por debajo de – 20°C
Deben ser manejados sobre la base de estudio del caso en particular (1,2).
3.8.7. Consideraciones adicionales para las condiciones de almacenamiento
Cuando por las características del PFT este no pueda mantenerse dentro del rango de temperatura y humedad de acuerdo con la zona climática IVb y sea destinado para almacenamiento en refrigerador, congelador o por debajo de -20ºC, siempre que se encuentre plenamente justificado, bibliográfica y experimentalmente con estudios de estabilidad de acuerdo con lo acá establecido, se aceptará la condición de temperatura y humedad que el titular sustente. En este caso, se debe garantizar que las condiciones de almacenamiento aprobadas por el Invima se conserven durante la vida útil del producto y se promueva que se mantengan durante el almacenamiento, distribución y uso del mismo.
Cuando por las características del PFT este requiera condiciones de temperatura y humedad diferentes a las ya mencionadas, el Invima evaluará las que el titular indique, siempre y cuando se presente la justificación, bibliográfica y experimental con los datos de soporte y los estudios de estabilidad. En este caso, el titular debe garantizar que las condiciones de almacenamiento aprobadas por el Invima se mantengan durante la vida útil del producto, el almacenamiento, distribución y uso del mismo
3.9. GARANTÍA DE LA ESTABILIDAD
Cuando los datos del estudio de estabilidad de largo plazo realizado sobre lotes primarios no cubran la vida útil propuesta en el momento de la solicitud, se deben presentar los respectivos soportes que garanticen la continuación y finalización de los estudios de estabilidad con posterioridad a la aprobación de la solicitud, con el fin de establecer, en forma segura, el periodo de vida útil del PFT en cuestión (1,2). La documentación correspondiente al proceso se debe entregar a la autoridad respectiva una vez terminado el estudio y también cuando lo requiera.
Cuando la solicitud incluya datos del estudio de estabilidad de largo plazo provenientes de lotes de producción, como se especificó en el numeral 3.4, que cubran la vida útil propuesta, no se considera necesario tomar una acción de garantía posterior a la aprobación. De lo contrario, se puede tomar una de las siguientes acciones (1):
a) Si la solicitud incluye datos provenientes del estudio de estabilidad, al menos sobre el número de lotes de producción, especificados en el numeral 3.4, pero no cubren el tiempo de vida útil propuesto, se debe continuar estos estudios a lo largo del periodo de la vida útil propuesta, contando con los estudios acelerados realizados por seis meses.
b) Si la solicitud incluye datos provenientes del estudio de estabilidad realizado sobre un número de lotes de producción inferior al especificado en el numeral 3.4, se debe continuar con estos estudios a lo largo del periodo de vida útil propuesto contando con los estudios acelerados por seis meses y colocar lotes de producción adicionales, hasta un total de por lo menos tres lotes, en estudios de estabilidad de largo plazo hasta cubrir el periodo de vida útil propuesto y el desarrollo de los estudios acelerados por seis meses.
c) Si la solicitud no incluye datos de estabilidad sobre lotes de producción, se debe iniciar el estudio de estabilidad de largo plazo con los primeros tres lotes de producción y se debe desarrollar hasta el periodo vida útil propuesto y el desarrollo de los estudios acelerados por seis meses.
El protocolo de estabilidad utilizado para los estudios de largo plazo, dentro de las actividades de garantía de estabilidad, debe ser el mismo que se estableció para los lotes primarios, a menos que científicamente se justifique otra cosa.
3.10. EVALUACIÓN
Se debe adoptar una aproximación sistemática para la presentación y evaluación de la estabilidad, la que debe incluir, cuando sea apropiado, resultados provenientes de los ensayos físicos, químicos, biológicos y microbiológicos, incluyendo atributos particulares de las formas de dosificación (p. ej. velocidad de disolución para las formas de dosificación sólidas orales) (1,2).
El propósito de los estudios de estabilidad es establecer, con base en el ensayo de un mínimo número de lotes del PFT, como se especificó en el numeral 3.4, la vida útil y las condiciones de almacenamiento para ser declaradas en la etiqueta, aplicables a todos los futuros lotes del PFT fabricados bajo circunstancias similares. El grado de variabilidad de los lotes individuales afecta la confiabilidad de que un lote futuro de producción permanezca dentro de las especificaciones a lo largo de su vida útil (1,2).
Cuando los datos presentan tan poca degradación y variabilidad que se puede concluir, de la observación de los datos, que el periodo de vida útil será confirmado, no sería necesario recurrir a la realización de un análisis estadístico para los estudios de largo plazo si se suministra la respectiva justificación (1,2). Este caso es más la excepción que la regla y la decisión definitiva sobre la necesidad de los cálculos se basa en la justificación presentada.
Una aproximación para el análisis de los datos de un atributo cuantitativo que se espera cambie con el tiempo, es determinar el momento en el que el límite de confianza del 95% de una sola cola (la inferior) para la curva promedio intercepta el criterio de aceptación. Si el análisis muestra que la variabilidad lote a lote es pequeña, es ventajoso combinar los datos y hacer una sola estimación global. Esto puede realizarse si se aplica primero un ensayo estadístico apropiado (por ejemplo valores de p para un nivel de significancia de rechazo de más de 0,25) a las pendientes de las líneas de regresión y a los interceptos a tiempo cero para los lotes individuales. Si no es apropiado combinar los datos de diferentes lotes, el periodo de vida útil global debe basarse en el tiempo mínimo al que un lote se espera que permanezca dentro del criterio de aceptación (1,2).
La naturaleza de cualquier relación de descomposición determinará, cuando los datos deben transformarse para un análisis de regresión lineal. Usualmente las relaciones pueden ser representadas por una función rectilínea, cuadrática o cúbica sobre una escala aritmética o logarítmica. Tanto como sea posible, la selección de un modelo debería justificarse por un razonamiento físico y/o químico y también tendría en cuenta la cantidad de datos disponibles (principio de parsimonia) para asegurar una predicción robusta. Los métodos estadísticos deben ser empleados para evaluar la bondad de ajuste de los datos de todos los lotes y los lotes combinados (donde sea apropiado) a la línea o curva de descomposición supuesta (1,2).
Si se justifica se podría extrapolar el límite de los datos provenientes del estudio en condiciones de largo plazo más allá del rango de observación, para extender el periodo de vida útil. Esta justificación debe estar fundamentada en lo que se conoce acerca del mecanismo de degradación, los resultados del ensayo bajo condiciones aceleradas, la bondad de ajuste a cualquier modelo matemático, tamaño de lote y la existencia de datos de estabilidad de soporte. Sin embargo, esta extrapolación supone que la misma relación de descomposición continuará aplicándose más allá de los datos observados (1,2).
Cualquier evaluación debe cubrir no solamente la valoración, sino también los niveles de los productos de descomposición y otros atributos apropiados. Donde sea conveniente, se debe poner atención a la revisión de la adecuación de la evaluación que une la estabilidad del producto farmacéutico terminado y el comportamiento de degradación durante el estudio (1,2).
3.11. LEYENDAS Y ETIQUETADO
Se debe establecer con base en la evaluación de la estabilidad del PFT, una leyenda para el almacenamiento con el fin de colocarla en la etiqueta. Donde sea aplicable se deben suministrar instrucciones específicas y particularmente para los PFTs que no pueden tolerar congelación o salidas de las condiciones de temperatura (1,2).
Debe existir coherencia entre la leyenda de las condiciones de almacenamiento indicadas en la etiqueta y la estabilidad demostrada del PFT (incluyendo lo concerniente a la estabilidad en uso, cuando aplica). Se debe colocar una fecha de expiración sobre la etiqueta del envase (1,2).
En principio, los PFTs deben estar empacados en envases que aseguren la estabilidad y protejan al PFT del deterioro. No se debe utilizar una leyenda de almacenamiento para compensar un envase inadecuado o inferior. Se podrían utilizar leyendas adicionales sobre la etiqueta en aquellos casos donde los resultados de los estudios de estabilidad demostraron factores limitantes (1,2).
3.12. ESTABILIDAD EN USO
El propósito de los estudios de estabilidad en uso es suministrar la información necesaria para el etiquetado de la preparación, las condiciones de almacenamiento y el periodo de utilización de los productos multidosis después de abierto el envase, la reconstitución o dilución de una solución, p.ej. un antibiótico para inyección suministrado como un polvo para reconstituir (1,2).
El ensayo debe ser diseñado tanto como sea posible, para simular el uso del PFT en la práctica, tomando en consideración el volumen de llenado del envase y cada vehículo empleado para la reconstitución y/o dilución en la forma indicada por el fabricante (si se indica más de un vehículo dispersante, se deben realizar ensayos con todos y cada uno de ellos). A intervalos de tiempo comparables, a aquellos que ocurren en la práctica, se deben remover cantidades apropiadas del producto por medio de métodos de muestreo comúnmente utilizados y descritos en la literatura del producto considerando sus características (1,2).
Las propiedades físicas, químicas y microbiológicas del PFT susceptibles de cambiar durante el almacenamiento, se deben determinar durante el periodo de vida útil de uso propuesto. Si es posible, los ensayos se deben desarrollar en puntos de tiempo inicial, intermedios y al final del periodo de vida útil de uso propuesto, sobre la cantidad final del PFT remanente en el envase. Se deben evaluar parámetros específicos como, por ejemplo, para el caso de los productos líquidos y semisólidos, sus preservantes tanto su contenido como efectividad (1).
Estos estudios se deben desarrollar en mínimo dos lotes, en un tamaño de escala por lo menos de lote piloto industrial durante el tiempo de uso propuesto, como parte fundamental de los estudios de estabilidad tanto al inicio y al final de la vida útil propuesta para el PFT. Si en el momento de la solicitud del Registro Sanitario no se cuenta con la totalidad de los datos a tiempo final de vida útil, los estudios del producto reconstituido o diluido se deben realizar al mes 12 o el último tiempo en evaluación a la fecha de solicitud. En general, no es necesario realizar nuevamente estos estudios en los lotes de garantía de estabilidad, cuando se hayan desarrollado hasta el tiempo final de la vida útil (1,2).
3.13. ESTUDIOS DE ESTABILIDAD EN LA ETAPA DE POS MERCADEO POR CAMBIOS QUE AFECTAN LA ESTABILIDAD
Una vez que el PFT ha sido registrado, se requerirán estudios de estabilidad adicionales, siempre que se hayan efectuado modificaciones que puedan afectar la estabilidad del IFA o del PFT (1). Se realizan inicialmente a condiciones aceleradas y posteriormente se ratifican con estudios de largo plazo. Dentro de las Buenas Prácticas de Manufactura, se puede considerar que estos estudios forman parte de las actividades de estandarización de los cambios efectuados.
A continuación, se enlistan algunas de las circunstancias bajo las cuales se deben adelantar este tipo de estudio (1):
a) Cambio cualitativo o cuantitativo de la formulación.
b) Cambio de fabricante del IFA, del PFT (incluyendo los cambios de lugar de las instalaciones o planta de un mismo fabricante).
c) Cambios en el proceso de manufactura del IFA o del PFT (incluyendo los cambios de equipos).
d) Cambio del envase primario y cierre del producto.
En todos los casos de modificaciones, los solicitantes deben investigar si el cambio efectuado tendrá o no un impacto sobre las características de calidad del IFA y el PFT y en consecuencia sobre su estabilidad (1). De acuerdo con lo anterior, es importante recordar que las modificaciones que se efectúen en el futuro y puedan afectar la estabilidad del IFA o del PFT conducen a la obsolescencia de las conclusiones del estudio de estabilidad inicial.
El alcance y diseño de los estudios de estabilidad para modificaciones y cambios está fundamentado en el conocimiento y experiencia adquirida sobre los IFAs y PFTs. Los resultados de estos estudios deben ser comunicados y autorizados por la entidad regulatoria (1).
3.14. ESTUDIOS DE ASEGURAMIENTO DE LA ESTABILIDAD (ON-GOING STABILITY)
Después de que ha sido otorgado el Registro Sanitario (autorización de comercialización), la estabilidad del PFT se debe monitorear de acuerdo con un programa continuo y apropiado, que permitirá la detección de cualquier evento de inestabilidad (p.ej. cambios en los niveles de impurezas o perfil de disolución) asociado con la formulación, en el sistema de envase y cierre en el que es comercializado. El propósito del programa de Aseguramiento de Estabilidad es monitorear el comportamiento del PFT y determinar que este permanece y puede permanecer dentro de las especificaciones bajo las condiciones de almacenamiento indicadas en la etiqueta, dentro del periodo de vida útil en todos los lotes futuros (1).
Esto aplica principalmente a los PFTs en el sistema de envase y cierre en el que fueron registrados, pero también se deben hacer algunas consideraciones para incluir los productos a granel, en el programa de estabilidad "on-going". Por ejemplo, cuando el producto a granel es almacenado por un periodo largo, antes de ser empacado y/o transportado de un sitio de manufactura a un sitio de envase; en este caso, se debe evaluar y estudiar el impacto de esta situación, sobre la estabilidad del producto empacado. Por lo general, este ensayo formaría parte de los estudios de estabilidad en la etapa de desarrollo, pero donde esta necesidad no ha sido prevista, se pueden suministrar los datos requeridos a la autoridad sanitaria mediante la inclusión de estas evaluaciones en el programa de estabilidad "on-going". También se puede aplicar consideraciones similares a los productos intermedios, que son almacenados y utilizados por periodos prolongados (1).
El programa de estabilidad "on-going" se debe describir en un protocolo escrito y los resultados se formalizan en un informe.
El protocolo para un programa de aseguramiento de estabilidad se debe extender hasta el final del periodo de vida útil y debe incluir, pero no estar limitado, a los siguientes parámetros (1):
a) Número de lote(s) por concentraciones y diferentes tamaños de lotes, si aplica. Si se emplean diferentes tamaños de lotes, se debe registrar el tamaño del lote.
b) Métodos de ensayo importantes: físicos, químicos, microbiológicos y biológicos.
c) Criterios de aceptación.
d) Referencias para los métodos de ensayo.
e) Descripción del sistema(s) de envase y cierre.
f) Frecuencia de ensayo (muestreo).
g) Descripción de las condiciones de almacenamiento (se deben utilizar condiciones estandarizadas para los ensayos de largo plazo como se describe en esta guía y consistentes con el etiquetado del producto).
h) Otros parámetros aplicables especificados para el PFT.
El protocolo para el programa de estabilidad "on-going", puede ser diferente al de los estudios iniciales de estabilidad de largo plazo, presentado cuando se sometió la documentación para la obtención del Registro Sanitario, siempre y cuando este cambio esté justificado y documentado en el protocolo (p.ej. la frecuencia de ensayos, o cuando se especifique que debe cumplir las recomendaciones revisadas) (1).
El número de lotes y la frecuencia de los ensayos deben suministrar datos suficientes para permitir evaluar la tendencia del análisis. Se debe incluir en el programa de estabilidad por lo menos al año, un lote de producción en cada concentración y tipo de envase primario, (a menos que ningún lote se produzca durante el año). Se puede aplicar el principio de diseño de extremos o matrices, si se justifica en el protocolo desde el punto de vista científico (1).
En ciertas situaciones se deben incluir lotes adicionales en el programa de estabilidad "on-going", tales como:
i) Cambio de fabricantes de excipientes siempre que se mantengan las mismas características (p.ej. hidratación, sales, ruta de obtención, envase, tamaño de partícula, etc.) de los excipientes iniciales.
ii) Cambio de escala de producción.
iii) Cambio de fabricante de los materiales de envase primario y cierre, manteniendo las mismas características iniciales (p.ej. tipo de material, tamaño, etc.).
iv) Reprocesamiento de productos.
v) Modificaciones en las instalaciones físicas y en los sistemas de apoyo crítico del área de fabricación del mismo fabricante.
Se deben investigar los resultados que se salen de las especificaciones o tendencias atípicas significativas. Se debe informar inmediatamente y por escrito a la autoridad sanitaria todo cambio significativo confirmado, los resultados que se salen de especificación o las tendencias atípicas significativas. La autoridad sanitaria evaluará el posible impacto sobre los lotes disponibles en el mercado (1).
Se debe escribir y mantener un resumen de todos los datos generados, incluyendo cualquier conclusión intermedia sobre el programa de estabilidad "on-going". Este resumen debe estar sujeto a revisiones periódicas (1).
Para todos los productos farmacéuticos terminados se debe establecer un programa rutinario de estudios de aseguramiento de la estabilidad, en condiciones de estabilidad natural, como una manera de establecer la verificación del cumplimiento de las Buenas Prácticas de Manufactura de los lotes que están en el mercado.
4. CONSIDERACIONES SOBRE EL DISEÑO DEL ESTUDIO DE ESTABILIDAD
Para el diseño de los estudios de estabilidad se debe tener en cuenta lo descrito a continuación:
4.1. ASPECTOS GENERALES
4.1.1. Metodología analítica indicadora de estabilidad
La metodología analítica, debe ser específica e indicadora de la estabilidad y debe estar validada o verificada (en caso de ser farmacopeica) por el fabricante del PFT o por el laboratorio que realiza el estudio de estabilidad, de acuerdo con lo indicado por la USP vigente y por la guía ICH Q2 (R1) (7). Se debe establecer con toda claridad cada uno de los parámetros evaluados, por ejemplo, especificidad, selectividad, precisión y exactitud del método (5,6,7). Esta información debe ser entregada junto con el estudio de estabilidad, con el detalle suficiente para permitir verificar la validación por parte de la autoridad sanitaria.
4.1.2. Temperaturas de almacenamiento
Se deben especificar las temperaturas reales de almacenamiento (numéricamente) utilizadas durante los estudios de estabilidad. Estas deben ser apropiadas y permanentemente registradas.
4.1.3. Calidad microbiológica
Los medicamentos que contienen agentes de conservación para controlar la contaminación microbiana deben tener un seguimiento del contenido de preservante(s), por lo menos al principio y al final del período de vida útil proyectado para el medicamento. Esta evaluación debe estar acompañada por el desarrollo de la prueba de eficacia antimicrobiana (1,2,5,6).
Las formas farmacéuticas estériles y no estériles que requieren control de la calidad microbiana, endotoxinas y esterilidad, deben ser evaluadas a intervalos específicos a lo largo del período de vida útil proyectado. Las pruebas deben realizarse al menos al principio y al final de la vida útil. Si los datos a largo plazo presentados a la autoridad reguladora para la solicitud del registro sanitario no cubren el período de vida útil completo, debe proporcionarse resultados de los ensayos al mes 12 o el último tiempo en evaluación a la fecha de solicitud (1).
4.1.4. Productos de descomposición o degradación
Cuando en la literatura se reporten productos de descomposición potencialmente riesgosos, provenientes del IFA, se debe entregar la siguiente información (5):
a) Identidad y estructura química (siempre que se encuentren definidas).
b) Resumen donde se referencia la información disponible acerca del efecto biológico de los productos de descomposición.
c) Especificaciones e instrucciones del ensayo para detectar la presencia de los productos de descomposición al nivel o concentraciones esperadas.
4.1.5. Consideraciones sobre los ensayos a efectuar según la forma farmacéutica en estudio
Los parámetros citados en el presente numeral, para cada una de las formas farmacéuticas se presentan como una guía para los tipos de pruebas que se pueden incluir en un estudio de estabilidad. En general, los ensayos de apariencia, valoración y degradación deben ser evaluados para todas las formas de dosificación, así como el contenido de preservante y antioxidante, si es aplicable (1).
De acuerdo con la forma farmacéutica en estudio, se deben evaluar las características de calidad en las que su alteración puede ser un signo importante de inestabilidad. Por ejemplo, el pH puede ser un ensayo indicador de estabilidad muy importante en algunas formas farmacéuticas, pero en otras carece de la mínima importancia.
Dentro de los ensayos a desarrollar se encuentra la valoración, la cual se usa para determinar la concentración del fármaco (o su contenido) en el PFT y es una prueba específica e indicadora de la estabilidad. Cuando se justifica una valoración no específica (p.ej. volumetría) se debe asegurar mediante otros procedimientos analíticos de sustento la capacidad de detectar cualquier especie interferente. Los resultados de la valoración a menudo se informan como porcentaje de la cantidad declarada, con criterios de aceptación que por lo general están en el intervalo de 90,0% a 110,0%, el cual toma en cuenta la variabilidad de la fabricación y la estabilidad durante la vida útil (6).
Según la evaluación del riesgo y las características individuales del IFA, la forma farmacéutica y el PFT, se debe establecer la frecuencia de los análisis.
Se deben desarrollar los ensayos necesarios dando cumplimiento a los requerimientos específicos de las características del IFA, la forma farmacéutica y el PFT. En caso de omisión de un ensayo específico, se debe proporcionar la respectiva justificación con el soporte científico, técnico y analítico para su evaluación. A continuación, se citan algunos ensayos según la forma farmacéutica:
4.1.5.1. Tabletas
Apariencia, friabilidad, dureza, color, humedad, impurezas, potencia y/o concentración, disolución (o desintegración, si se justifica) y calidad microbiológica (1,5,6,8).
4.1.5.2. Cápsulas
Potencia y/o concentración, impurezas, humedad, color, apariencia, forma, disolución (o desintegración, si se justifica) y calidad microbiológica. Para las cápsulas blandas de gelatina, el contenido debe examinarse para ver la formación de precipitado, turbidez, fugas, formación de películas y pH, cuando aplique (1,5,6,8).
4.1.5.3. Emulsiones
Apariencia (tal como separación de fases), color, pH, viscosidad, impurezas, calidad microbiológica, tamaño medio y distribución de los glóbulos dispersados y potencia y/o concentración. Se recomienda un almacenamiento colocando los envases en posición de pie e invertida o lateral, para evaluar el contacto con el sistema de cierre. Además, el empleo de ciclos de calentamiento y enfriamiento como, por ejemplo, entre 4ºC y 45ºC (1,5,6,8).
4.1.5.4. Soluciones y suspensiones orales
Apariencia (formación de precipitado y turbidez), potencia y/o concentración, impurezas, pH, color, calidad microbiológica. Adicionalmente, para las soluciones, claridad y para las suspensiones redispersabilidad, propiedades reológicas, disolución y tamaño medio y distribución de las partículas. Se recomienda el almacenamiento de las preparaciones líquidas y las suspensiones en forma lateral o invertida, con el fin de determinar cuando el contacto del medicamento con el sistema de cierre afecta la integridad del producto (1,5,6,8).
4.1.5.5. Polvos y gránulos orales para reconstituir antes del uso
Apariencia, concentración, color, impurezas, calidad microbiológica y humedad en el producto en polvo (1,5,6,8).
El medicamento reconstituido (soluciones y suspensiones) debe ser preparado como se indica en la etiqueta y las características a evaluar en el producto reconstituido hasta el período máximo de uso previsto pueden ser: Apariencia, pH, dispersabilidad, calidad microbiológica y potencia y/o concentración (1,5,6,8).
En estos productos para reconstituir se deben desarrollar dos estudios de estabilidad, el primero ampara el almacenamiento del producto hasta el momento de su reconstitución y uso y el segundo ampara el periodo de tiempo durante el que el producto puede ser utilizado después de haber sido reconstituido de la manera indicada y mantenido en las condiciones recomendadas (1,2).
4.1.5.6. Dosificadores de aerosoles para inhalación
Para todos los tamaños de envase primario/cierre: Concentración, impurezas, uniformidad de contenido de la dosis, número de dosis medidas que cumplen la uniformidad de contenido, color, claridad en las soluciones, distribución de tamaño de partícula en las suspensiones, pérdida del propelente, presión, evaluación microscópica, contenido de agua, calidad microbiológica, corrosión de la válvula y patrón de rocío, tasa de fugas, suministro de válvulas (peso de la inyección), extraíbles/lixiviables de componentes plásticos y elastoméricos, suministro de bomba, partículas extrañas y extraíbles y componentes elastómeros del recipiente, cierre y bomba. Las muestras se deben almacenar de pie e invertida o lateral (1,5,6,8).
Como el contenido de los recipientes está bajo presión, el llenado de los envases debe ser verificado por pérdida de peso durante todo el período de vida útil del producto. Para las suspensiones, la formación de agregados o solvatos puede producir la obstrucción de las válvulas o la liberación de una dosis farmacológicamente inactiva. La corrosión de la válvula de dosificación o el deterioro del empaque puede afectar adversamente la liberación de la cantidad correcta de IFA (1).
4.1.5.7. Sprays nasales (soluciones y suspensiones)
Claridad (para soluciones), calidad microbiológica, pH, impurezas, material particulado, patrón de rocío, uniformidad de contenido de la dosis, número de dosis medidas que cumplen la uniformidad de contenido, concentración, distribución de las gotas y/o tamaño de partícula, pérdida de peso, suministro de la bomba, evaluación microscópica (para suspensiones), partículas extrañas y extraíbles/lixiviables de los componentes de plástico y elastómeros del recipiente, cierre y bomba (1,6). Se recomienda un almacenamiento colocando los envases en posición de pie e invertida o lateral, para evaluar el contacto con el sistema de cierre.
4.1.5.8. Preparaciones tópicas, óticas y oftálmicas
En productos tópicos como ungüentos, cremas, lociones, soluciones, geles, gotas para los ojos y sprays cutáneos, deben examinarse las siguientes características:
a) Preparaciones tópicas: Apariencia, claridad, homogeneidad, pH, impurezas, suspensibilidad (para lociones), consistencia, viscosidad, distribución de tamaño de partícula (para suspensiones cuando sea posible), concentración, calidad microbiológica/esterilidad y pérdida de peso/volumen extraíble cuando sea apropiado (1,6,8).
b) En las preparaciones oftálmicas u óticas (p. ej. Soluciones, suspensiones, cremas y ungüentos) adicional se debe realizar: Apariencia, consistencia, pH, esterilidad, material particulado, volumen extraíble (1,6,8).
c) La evaluación de los sprays cutáneos debe incluir: presión, pérdida de peso, peso neto dispensado, velocidad de suministro, calidad microbiológica, patrón de rocío, concentración, impurezas, contenido de agua y distribución del tamaño de partícula (para suspensiones) (1,6,8).
Para estas preparaciones, cuando aplique, se recomienda un almacenamiento colocando los envases en posición de pie e invertida o lateral, para evaluar el contacto con el sistema de cierre.
4.1.5.9. Parenterales de pequeño volumen
Los parenterales de pequeño volumen incluyen un rango extremadamente amplio de preparaciones y envases primarios/cierres, todo lo que debe ser incluido en el estudio de estabilidad. La evaluación de estos medicamentos debe incluir por lo menos las siguientes características: Concentración y/o potencia, impurezas, apariencia, color, claridad (para soluciones), material particulado, pH, esterilidad y endotoxinas (a intervalos razonables), por lo menos al inicio y al final del estudio (1,6,8).
Los estudios de estabilidad de productos en polvo para reconstituir a solución inyectable, deben incluir color, tiempo de reconstitución y contenido de agua. Los parámetros específicos que deben ser examinados a intervalos apropiados hasta completar el período de máximo tiempo de empleo indicado para el producto reconstituido y almacenado bajo las condiciones recomendadas en la etiqueta, deben incluir: Apariencia, color, pH, concentración y/o potencia, dispersabilidad, material particulado, claridad, esterilidad y pirógenos/ endotoxinas. Se considera apropiado monitorear la esterilidad después de la reconstitución de un producto en el que se indica que la reconstitución se realiza sin comprometer la esterilidad (p.ej. jeringas de doble cámara) (1,6,8).
En las suspensiones para inyección se debe incluir adicional a lo mencionado, la distribución del tamaño de partícula, dispersibilidad y propiedades reológicas y en emulsiones incluir la separación de fases, viscosidad, distribución y tamaño promedio de los glóbulos en la fase dispersa (1,6,8).
En los liposomas para inyección, según corresponda, se debe incluir adicional a lo mencionado, la composición de lípidos, ácidos grasos y productos de degradación (lisolípidos), distribución del tamaño de partícula, laminalidad, porcentaje de lípidos libres vs porcentaje de lípidos encapsulados, fármaco libre vs Fármaco encapsulado (6).
Los productos parenterales, con excepción, por ejemplo, de ampollas y bolsas, deben ser almacenados en forma invertida o sobre un costado, con el fin de determinar por comparación, cuando el contacto del medicamento o del solvente con el sistema de cierre afecta la integridad del producto o resulta en una extracción de los compuestos químicos por el material del sistema de cierre (1).
4.1.5.10. Parenterales de gran volumen
Los ensayos de estabilidad para parenterales en gran volumen son similares a aquellos indicados para los parenterales en pequeño volumen.
Una evaluación mínima debe incluir los siguientes aspectos: concentración, apariencia, color, claridad, material particulado impurezas, pH, volumen y detección de materiales extractables cuando aplica, esterilidad y pirógenos/endotoxinas (1,6,8).
Estos productos deben ser almacenados, a excepción por ejemplo de bolsas, colocando algunos recipientes en forma invertida y lateral, con el fin de determinar cuando el solvente o el producto en contacto con el sistema envase/cierre afecta la integridad del producto o conduce a la extracción de compuestos químicos a partir del material del sistema envase/ cierre (1).
4.1.5.11. Supositorios y óvulos
Se debe evaluar la potencia y/o concentración, intervalo de temperatura de ablandamiento, apariencia, impurezas, calidad microbiológica y disolución/desintegración (cuando corresponda) a 37°C (1,6,8).
4.1.5.12. Sistemas transdérmicos
Apariencia, liberación por área superficial (velocidad de liberación in vitro), potencia y/o concentración, impurezas, prueba de fugas, adherencia, desprendimiento, disolución, calidad microbiológica/esterilidad (1,6,8).
4.1.5.13. Mezclas de medicamentos (PFTs)
Todo medicamento que se destina a ser utilizado como un aditivo de otro medicamento, tal como ocurre en las mezclas para administración parenteral y enteral, existe la posibilidad del desarrollo de incompatibilidades. En tal situación, aquel medicamento en cuya etiqueta se indica que es para ser administrado previa adición sobre otro medicamento (parenterales, aerosoles) se debe evaluar la estabilidad y la compatibilidad en la mezcla con los otros medicamentos.
Se sugiere un protocolo de estudio de estabilidad que establezca pruebas a realizar a los siguientes intervalos: 0, 6, 8 y 24h o cualquier otro esquema apropiado al tiempo de utilización del producto. Las pruebas a realizar deben comprender los siguientes aspectos (5):
a) Evaluación completa del medicamento portador y del medicamento adicionado.
b) Valoración de los diferentes principios activos en la mezcla.
c) pH (en especial para los parenterales en gran volumen no bufferizados), color, claridad, apariencia.
d) Material particulado.
e) Interacción con el envase.
4.1.5.14. Dispositivos intrauterinos y vaginales considerados como medicamentos
Si el dispositivo contiene un reservorio del principio activo, a partir del que, el fármaco se difunde a través de una membrana de control de liberación o algún tipo de matriz polimérica que contiene un principio activo uniformemente disperso dentro de esta, se debe evaluar el contenido total del IFA, detectar la presencia de productos de descomposición y medir la velocidad de liberación del IFA "in vitro", y la esterilidad del dispositivo (5).
4.1.5.15. Otras formas farmacéuticas
Para formas farmacéuticas no especificadas en esta sección, cada caso será revisado particularmente. Los ensayos requeridos para el PFT, deben estar de acuerdo con lo dispuesto en las respectivas monografías o para la forma farmacéutica en las Farmacopeas oficiales. Cuando el PFT no es oficial en las Farmacopeas aceptadas en Colombia se deben realizar los ensayos de acuerdo con los requerimientos específicos de las características del IFA, la forma farmacéutica y el PFT.
4.1.6. Consideraciones de tipo estadístico
Un protocolo de estudio de estabilidad debe describir no solamente, cómo se ha diseñado y llevado a cabo el estudio de estabilidad, sino también los métodos estadísticos a ser utilizados en el análisis de los datos.
4.2. ASPECTOS ESPECÍFICOS
4.2.1. Consideraciones para el diseño experimental
Se debe tener en cuenta, la variabilidad individual entre las unidades de dosificación, la variabilidad de los recipientes dentro de un lote y la variabilidad de los lotes entre sí, con el fin de asegurar que los datos resultantes para cada lote, sean realmente representativos del lote como un todo y sirvan para cuantificar la variabilidad de lote a lote. El grado de variabilidad afecta la confianza que se pueda tener en la probabilidad de que un futuro lote permanezca dentro de las especificaciones hasta su fecha de expiración (5).
4.2.2. Consideraciones a aplicar para el muestreo
Durante el desarrollo de un estudio de estabilidad se debe muestrear a diferentes tiempos y cada muestreo estará conformado por un número de muestras apropiado, en donde se considera que tres muestras independientes es el valor mínimo aceptable. El tipo de muestra a utilizar dependerá de la clase de forma farmacéutica y en especial de su homogeneidad, es así como se puede hacer uso de muestras simples y compuestas (5).
La selección de los envases (frascos, ampollas, viales, blister, tiras, etc.) a partir de los lotes escogidos para el estudio, se debe llevar a cabo de tal manera que se asegure que las muestras sean representativas del lote en estudio. Esto se logra mediante la toma al azar de una muestra representativa de los envases que conforman el lote, utilizando un plan en donde se comienza en un punto al azar y a partir de éste se toma el enésimo recipiente de cada fila (n muestras son tomadas de la totalidad del lote). También se puede emplear cualquier otro plan de muestreo que garantice una selección no sesgada de las muestras (5).
Las muestras a ser evaluadas en un tiempo de muestreo determinado deben ser tomadas de envases no abiertos con anterioridad. Por esta razón, deben colocarse en estudio tantos envases como sea necesario según los tiempos de muestreo establecidos en el estudio de estabilidad y el tipo de muestra a utilizar (simple o compuesta). En cualquier caso, se debe muestrear por lo menos tres muestras independientes para cada tiempo de muestreo (5).
Se debe tener como regla que las unidades de dosificación tomadas a partir de un envase dado, deben ser muestreadas al azar, de manera que cada unidad de dosificación tenga la misma probabilidad de ser incluida en la muestra. Si se cree que las unidades entran al envase al azar, entonces se acepta el muestreo de las unidades de la parte superior, una vez se abra el envase. Es importante recordar que el procedimiento de muestreo forma parte integral de los protocolos de los estudios de estabilidad (5).
Si más de un recipiente se muestrea en un tiempo de muestreo dado, se puede combinar dentro de la muestra compuesta un número igual de unidades de cada recipiente para conformar cada muestra independiente. Se recomienda que el mismo tamaño de muestra compuesta sea utilizado a través de todo el estudio de estabilidad (5).
Si se utiliza la técnica de muestras compuestas, su conformación se debe describir en el protocolo del estudio de estabilidad (5).
4.2.3. Consideraciones sobre el número de muestras a utilizar
El número de unidades o cantidad por muestra a tomar, para cada uno de los ensayos a realizar (p.ej. disolución, uniformidad de dosis, material particulado, microbiológico, etc.), debe estar de acuerdo con lo indicado en las Farmacopeas aceptadas en Colombia y en la técnica del laboratorio fabricante o del laboratorio que realiza el estudio de estabilidad.
Se debe mantener independencia en el muestreo de cada tipo de envase y en cualquier otra diferencia existente entre los lotes, puesto que las conclusiones son individuales para un producto en cuestión.
Se recomienda el siguiente número de muestras para la cuantificación (valoración) del(los) IFA(s) en cada tiempo de muestreo, como el mínimo estadísticamente aceptable por lote de producto utilizado. En el caso que se justifique científica, estadística y técnicamente, es posible emplear otras alternativas (5,6):
a) Para sólidos dosificados en envase y/o empaque individualizado: como tabletas, cápsulas, grageas, óvulos, supositorios, etc. en blíster, sobres o tiras; se tomarán mediante un plan de muestreo al azar predefinido, 3 muestras compuestas independientes de cada lote, en cada tiempo de muestreo, las que se deben valorar individualmente.
b) Para sólidos dosificados en envase primario de multidosis como: tabletas, cápsulas, grageas, etc., en frascos o envases primarios que contienen 10 o más unidades, tomar mediante un plan de muestreo al azar predefinido, 3 envases de cada lote y extraer las unidades requeridas para realizar el análisis de 3 muestras compuestas independientes, en cada tiempo de muestreo, las que se deben valorar individualmente.
c) Polvos o granulados dosificados en sobres o en sachets: tomar mediante un plan de muestreo al azar predefinido el número de sobres correspondiente para tres muestras simples o compuestas independientes (según corresponda), en cada tiempo de muestreo, las que se deben valorar individualmente.
d) Polvos o granulados a granel (multidosis): en presentación por tarros o frascos, se toman mediante un plan de muestreo al azar predefinido 3 tarros o frascos, de cada uno de los envases se extrae una muestra simple de la parte superior y una muestra simple de la parte inferior, para un total de 6 muestras, las cuales se deben valorar individualmente. El tamaño de la muestra corresponde al requerido para realizar los análisis, dependiendo de la concentración de los principios activos.
e) Sólidos para reconstituir antes del uso en empaque unidosis y multidosis, manejar cada caso como se indica en el literal C.
f) Productos líquidos en envase unidosis: para ampollas, tomar mediante un plan de muestreo al azar predefinido el número de unidades correspondientes para obtener tres muestras compuestas independientes, en cada tiempo de muestreo, las que se deben valorar individualmente para frascos viales, tomar mediante un plan de muestreo al azar predefinido, 6 frascos viales por cada lote, tres colocados en posición normal (de pie) y otros tres en posición invertida o lateral, para que el producto quede en contacto con el sistema de cierre, de cada frasco se toma una muestra que debe ser valorada en forma independiente.
g) Productos líquidos en envase multidosis como: jarabes, elixires, soluciones, suspensiones, emulsiones, etc., seguir lo indicado para frascos viales (literal f).
h) Productos semisólidos en envase unidosis y multidosis como: cremas, ungüentos, geles, etc., seguir lo indicado a continuación en las presentaciones de tubos o tarros, se toman mediante un plan de muestreo al azar predefinido 3 envases, de cada uno de ellos se extrae una muestra simple, las cuales se deben valorar individualmente. El tamaño de la muestra corresponde al requerido para realizar los análisis, dependiendo de la concentración de los principios activos.
Para realizar el ensayo de uniformidad de contenido proceder de la siguiente manera:
Para productos envasados en tubos que contienen 5 g o más, tomar mediante un plan de muestreo al azar predefinido 3 unidades. De cada uno de los tubos se extrae una cantidad apropiada del producto de la parte superior, media e inferior, para un total de 9 muestras, las cuales se deben valorar individualmente.
Para productos envasados en tubos que contienen menos de 5 g, tomar mediante un plan de muestreo al azar predefinido 3 unidades. De cada uno de los tubos se extrae una cantidad apropiada del producto de las partes superior e inferior, para un total de 6 muestras, las cuales se deben valorar individualmente.
Para los productos en envases diferentes a los tubos, para los que no se pueda usar el método de muestreo anteriormente presentado, se pueden aceptar otros métodos como el descrito a continuación para un tarro:
- Seleccionar una jeringa adecuada cuya longitud sea suficiente para alcanzar el fondo del recipiente.
- Retirar el émbolo de la jeringa y cortar la parte inferior del cuerpo de la jeringa. El muestreo se debe llevar a cabo en un punto a la izquierda o la derecha de una línea imaginaria que divida a la mitad la superficie del tarro, a fin de conservar una región intacta en el otro lado para cualquier investigación adicional (Ver la Figura 1).
- Presionar lentamente el cuerpo de la hacia el interior del envase hasta alcanzar el fondo. Luego, hacer girar el cuerpo de la jeringa que contiene el núcleo de la muestra y retirar la jeringa del envase.
Insertar el émbolo de la jeringa en el cuerpo y extrudir cuidadosamente el núcleo de la muestra sobre una superficie limpia en tres porciones iguales que representen a las partes superior, media e inferior del envase. De cada una de estas porciones tomar una muestra adecuada, para un total de 9 muestras, las cuales se deben valorar individualmente

Figura 1. Muestreo de un tarro.
i) Para formas farmacéuticas no especificadas en esta sección, cada caso será revisado particularmente. Los planes de muestreo, el número de envases y las cantidades requeridas para la muestra, deben estar de acuerdo con lo dispuesto en las respectivas monografías o para la forma farmacéutica en las Farmacopeas aceptadas en Colombia y en la técnica del laboratorio fabricante o del laboratorio que realiza el estudio de estabilidad. Tener en cuenta que en cada tiempo de muestreo se deben emplear como mínimo tres muestras independientes que deben ser analizadas individualmente por lote evaluado.
5. ANÁLISIS DE LOS DATOS E INTERPRETACIÓN DE LOS ESTUDIOS DE ESTABILIDAD (9)
Los análisis e interpretación de los estudios de estabilidad se requieren para establecer, con un alto grado de confianza, un período de vida útil durante el cual el promedio de las características (por ejemplo, la potencia y/o concentración) del lote de producto permanece dentro de las especificaciones. Este período de vida útil debe ser aplicable a todos los futuros lotes producidos por medio del mismo proceso de manufactura establecido para el IFA o PFT. No es suficiente el que un período de vida útil propuesto asegure que el proceso promedio está dentro de las especificaciones, es indispensable considerar las variaciones de los lotes en forma individual, de manera que, al final del tiempo de vida útil todas las muestras garanticen que los lotes se encuentran dentro de especificaciones.
Aunque se esperan variaciones normales de fabricación y procesos analíticos, es importante que el producto farmacéutico se formule con la intención de proporcionar el 100% de la cantidad etiquetada del IFA en el momento de la liberación del lote. Si los valores de ensayo de los lotes utilizados para soportar la solicitud de registro son superiores al 100% de lo indicado en la etiqueta en el momento de la liberación del lote, se puede sobrestimar la vida útil propuesta en la solicitud. Por otra parte, si el valor de ensayo de un lote es inferior al 100% de lo indicado en la etiqueta en el momento de la liberación del lote, podría caer por debajo del criterio de aceptación inferior antes del final de la vida útil propuesta.
Debe adoptarse un enfoque sistemático en la presentación y evaluación de la información sobre estabilidad. La información de estabilidad debe incluir, según proceda, los resultados de las pruebas físicas, químicas, biológicas y microbiológicas, incluidas las relacionadas con atributos particulares de la forma farmacéutica (p.ej. la disolución). Se debe evaluar la adecuación del balance de masa. Se deben considerar los factores que pueden causar una aparente falta de balance de masa incluyendo, por ejemplo, los mecanismos de degradación y la capacidad indicadora de estabilidad y la variabilidad inherente de los procedimientos analíticos.
Los datos de los estudios de estabilidad y, cuando proceda, los datos de soporte, deben evaluarse para determinar los atributos críticos que puedan influir en la calidad y el desempeño del IFA o el PFT. Cada atributo debe evaluarse por separado y debe hacerse una evaluación general de los hallazgos con el fin de proponer un período de re análisis o de vida útil. El periodo de vida útil propuesto no debe exceder el predicho para ningún atributo individual.
El árbol de decisiones del Apéndice 1 describe un enfoque escalonado para la evaluación de los datos de estabilidad, así como cuándo y cuánto se puede considerar la extrapolación para una vida útil propuesta. Una extrapolación de los datos de estabilidad presupone que el mismo patrón de cambio continuará aplicándose más allá del período cubierto por los datos a largo plazo. La corrección del supuesto patrón de cambio es crítica cuando se considera la extrapolación. Una vida útil concedida sobre la base de la extrapolación, siempre debe verificarse mediante datos adicionales de estabilidad a largo plazo tan pronto como estos datos estén disponibles.
Para los IFAs o los PFTs destinados a ser almacenados bajo las condiciones de la Zona climática establecida, la evaluación debe comenzar con cualquier cambio significativo en la condición acelerada y progresar a través de las tendencias y la variabilidad de los datos a largo plazo. En el apéndice 1 se proporciona un árbol de decisión para este fin.
Cuando no se produzca un cambio significativo en la condición acelerada, el período de vida útil dependerá de la naturaleza de los datos a largo plazo y acelerados.
Cuando se produce un cambio significativo en la condición acelerada, el período de vida útil dependerá del resultado de los estudios de estabilidad en la condición a largo plazo.
Las condiciones estándar para el análisis de datos e interpretación de los estudios de estabilidad y los ejemplos de enfoques estadísticos para el análisis de datos (descritos en la guía ICH Q1E) pueden ser consultadas en la página web del Invima en el apartado de estabilidad, con sus respectivas actualizaciones.
En caso de emplear procedimientos estadísticos, para la estimación del período de re análisis o vida útil, distintos a los descritos en la página web mencionada, se debe presentar la información correspondiente donde se describa de forma detallada el procedimiento desarrollado, junto con la debida justificación, para ser evaluado por la autoridad regulatoria.
6. ESTUDIOS DE ESTABILIDAD ESPECIALES
Un programa de estabilidad no solo cubre los estudios realizados en el marco del registro sanitario, sino que también incluye estudios que se crean para proporcionar datos de apoyo a otros programas tales como almacenamiento a granel, producto en proceso o intermedio, excursiones, etc. Este capítulo ofrece información de estos y otros estudios para apoyar estos propósitos.
6.1. PARA APOYAR EXCURSIONES DE LAS CONDICIONES DE ALMACENAMIENTO Y DISTRIBUCIÓN
Los controles ambientales juegan un papel clave en el mantenimiento de la seguridad, calidad y eficacia de los medicamentos. Los PFTs se deben almacenar y distribuir de acuerdo con las condiciones de almacenamiento predeterminadas (por ejemplo, temperatura) respaldadas por datos de estabilidad. Las excursiones de temperatura fuera de sus respectivas condiciones de almacenamiento establecidas, por períodos breves, pueden ser aceptables siempre que los datos de estabilidad y la justificación científica / técnica demuestre que la calidad del producto no se ve afectada. Los fenómenos de excursión de temperatura también son aplicables durante el almacenamiento y distribución de los IFAs antes de su recepción en el sitio de fabricación del PFT.
Se pueden utilizar los datos provenientes de las condiciones aceleradas de almacenamiento para evaluar el efecto de las desviaciones de corta duración por fuera de las condiciones de almacenamiento etiquetadas, durante la distribución como fueron descritas en los numerales 2.7, 3.8 y 5 (1,2,9). Puede ser necesario realizar estudios complementarios como los estudios de ciclos de temperatura (térmicos, refrigeración, congelación), diseñados para evaluar las fluctuaciones de las condiciones ambientales que pueden ocurrir durante la distribución del PFT (6,10,11,12,13,14). Se pueden utilizar otras alternativas con el debido sustento científico y técnico.
6.2. PRODUCTO A GRANEL, EN PROCESO O INTERMEDIO
Los estudios de estabilidad deben realizarse para apoyar el almacenamiento de producción y envase. Cuando el producto a granel es almacenado por un periodo largo de tiempo, antes de ser envasado y/o transportado de un sitio de manufactura a un sitio de envase, se debe evaluar y estudiar el impacto de este proceso sobre la estabilidad del producto. Por lo general, este ensayo formaría parte de los estudios de estabilidad en la etapa de desarrollo, pero donde esta necesidad no ha sido prevista, se pueden suministrar los datos requeridos a la autoridad sanitaria mediante la inclusión de estas evaluaciones en el programa de estabilidad "on-going" (1).
Comúnmente estos productos se almacenan en un envase, como bolsa de polietileno doble en un tambor o en recipientes de plástico, con el fin de imitar el envase del producto a granel. En estos casos se deben realizar estudios en por lo menos un lote industrial cuando el estudio se desarrolle dentro del programa de estabilidad "on-going". Se recomienda que las pruebas críticas se realicen cada 3 meses; en el caso de manejar otras frecuencias, estas no deben sobrepasar los 6 meses. También se pueden aplicar consideraciones similares a los productos intermedios y en proceso (p.ej. mezcla de polvos previa compresión), que son almacenados y utilizados por periodos prolongados (1).
Estos estudios se realizan una única vez, siempre y cuando se mantengan por completo las condiciones de almacenamiento evaluadas y establecidas en el estudio.
7. PRESENTACIÓN DEL PROTOCOLO E INFORME DEL ESTUDIO DE ESTABILIDAD
Se recomienda que el protocolo e informe del estudio de estabilidad incluyan la siguiente información y datos, para facilitar las decisiones relacionadas con la aceptación del estudio de estabilidad y la vida útil propuesta. En caso de ausencia de alguno de los aspectos solicitados se debe presentar la justificación técnica al respecto.
7.1. ELEMENTOS DEL INFORME DEL ESTUDIO DE ESTABILIDAD DEL IFA
a) Nombre del IFA (en Denominación Común Internacional).
b) Fabricante del IFA, envasador y/o Acondicionador si aplica y lugar donde se realiza el estudio de estabilidad.
c) Descripción general del proceso de fabricación del IFA.
d) Objetivo del estudio de estabilidad.
e) Tipo de estudio de estabilidad y condiciones de almacenamiento.
f) Número, tamaño y fecha de fabricación de los lotes.
g) Descripción del material de envase (s), cierre y empaque (si aplica).
h) Duración del estudio, frecuencia y tiempos de muestreo.
i) Especificaciones y criterios de aceptación.
j) Resultados de los ensayos bajo stress.
k) Resultados de las pruebas desarrolladas y descripción del método analítico utilizado, según corresponda.
l) Vida útil y condiciones de almacenamiento.
m) Condiciones recomendadas para las excursiones y la distribución.
n) Análisis de resultados y conclusiones del estudio.
7.2. ELEMENTOS DE UN PROTOCOLO DE ESTUDIOS DE ESTABILIDAD DEL PFT
El protocolo para un estudio de estabilidad se debe prolongar hasta el final del periodo de vida útil y debe incluir, pero no está limitado a los siguientes parámetros:
a) Nombre del IFA (en Denominación Común Internacional) y del PFT.
b) Titular del Registro Sanitario e Importador (según corresponda)
c) Fabricante del IFA, producto intermedio y/o PFT, envasador y/o Acondicionador si aplica.
d) Objetivo del estudio de estabilidad
e) Lugar donde se realiza el estudio de estabilidad
f) Laboratorio de análisis quien realiza el estudio de estabilidad
g) Tipo de estudio de estabilidad
h) Condiciones de almacenamiento del estudio de estabilidad
i) Número y tamaño de lotes del IFA o PFT a evaluar, incluyendo fecha de fabricación
j) Descripción de la forma farmacéutica y de la presentación comercial evaluada
k) Duración de los estudios de estabilidad
l) Frecuencia y tiempos de muestreo
m) Fórmula cualicuantitativa del PFT o producto intermedio (según corresponda)
n) Especificaciones del IFA o PFT (fisicoquímicas y microbiológicas)
o) Descripción del sistema de envase (s) y cierre
p) Descripción del material de empaque
q) Ensayos a realizar por cada uno de los tiempos
r) Especificaciones y metodología a desarrollar para producto reconstituido/diluido.
s) Descripción del número de unidades a utilizar por cada muestra y ensayo en cada tiempo de muestreo y el número total de unidades ingresadas al estudio.
t) Cuando aplique, descripción de la orientación de las muestras (de pie, invertido, etc.) para cada lote evaluado.
u) Número, código y/o nombre del método analítico validado (según corresponda)
v) Información de los estándares utilizados
w) Diseño experimental propuesto.
7.3. ELEMENTOS DEL INFORME DEL ESTUDIO DE ESTABILIDAD DEL PFT
El informe para un estudio de estabilidad debe incluir, pero no está limitado a lo descrito para el protocolo en el numeral 7.2 y entre otros, adicionar los siguientes parámetros:
a) Número de unidades de dosificación seleccionadas y aclarando si los ensayos fueron realizados sobre unidades individuales o sobre compuestos de unidades individuales (muestra compuesta).
b) Se debe detallar lo referente a la orientación de las muestras (de pie, invertido, etc.) para cada lote evaluado.
c) Metodología estadística aplicada al muestreo, tratamiento y análisis de los datos y resultados.
d) Fechas de muestreo y análisis.
e) Los datos obtenidos (p.ej. en valoración o potencia, disolución, impurezas, etc.) deben ser presentados en forma tabulada individual. También deben ser tabulados los promedios resultantes de los datos, con los límites de confianza del 95%, debidamente identificados.
f) Anexar la documentación sobre los métodos estadísticos apropiados y las fórmulas utilizadas en el análisis de los datos y resultados intermedios.
g) Anexar la evaluación de los datos, incluyendo los cálculos, análisis estadísticos y gráficas.
h) Especificaciones para la liberación del IFA o PFT de acuerdo con los resultados obtenidos en el estudio de estabilidad.
i) Señalar la vida útil propuesta y su justificación correspondiente.
j) Análisis de tendencia de los datos.
k) Análisis técnico detallado de resultados con cambios significativos, acciones preventivas, correctivas y conclusiones al respecto.
l) Análisis técnico detallado de resultados fuera de especificaciones, acciones preventivas, correctivas y conclusiones al respecto.
m) Análisis y evaluación de los efectos de las excursiones que excedan las tolerancias definidas por más de 24 horas. Acciones preventivas, correctivas y conclusiones al respecto
n) Definición de las condiciones de almacenamiento para el IFA, PFT o PFT reconstituido según corresponda.
o) Condiciones recomendadas para la distribución.
p) Conclusiones finales del estudio de estabilidad.
q) Datos y soportes de los análisis realizados según corresponda.
r) Observaciones y recomendaciones finales.
Los registros e informes de los estudios de estabilidad deben revisarse en periodos de tiempo definidos y estar disponibles para inspecciones en cualquier momento. Los registros de laboratorio incluyen un registro completo de datos y descripción de muestras tales como almacenamiento, ubicación, cantidad, lote, fecha de recepción, etc. Los datos de estabilidad deben ser trazables y tener los soportes que los sustenten en caso de inspecciones por la entidad regulatoria.
8. GLOSARIO
Aerosol: Forma farmacéutica que consta de una preparación líquida o sólida envasada a presión y destinada para administración en forma de fino rocío. El término descriptivo "aerosol" también hace referencia al fino rocío de gotitas o partículas sólidas emitidas por el producto.
Balance de masa: El proceso de sumar el valor de la valoración y los niveles de los productos de degradación para ver cuán estrechamente se suman al 100% del valor inicial, teniendo en cuenta el margen de error analítico. Idealmente, cuando ocurre la degradación, la perdida en el ensayo de valoración del medicamento debe correlacionarse bien con el aumento medido en los productos de degradación.
Cambio en el tiempo y variabilidad: Se refiere a las variaciones en los resultados de los ensayos dentro del intervalo de análisis establecido, las cuales son inferiores a lo descrito para cambios significativos. Se considera poco cambio en el tiempo y poca variabilidad aquellos resultados que se mantienen muy cercanos al resultado inicial y cambio en el tiempo y/o la variabilidad, aquellos resultados que se encuentran dentro del intervalo establecido entre el resultado inicial y el límite considerado como cambio significativo.
Cierre: Sistema o dispositivo que impide la salida del contenido de un envase determinado, o el ingreso de cualquier material extraño y es parte constitutivo del envase.
Componentes del envase: Cualquier parte del envase o del sistema de envase-cierre, incluyendo el envase primario (p.ej., ampollas, jeringas prellenadas, viales, frascos); el recubrimiento interno del envase primario (p.ej., recubrimiento interno en cartucho tubular); el cierre (p.ej., tapas de rosca, tapones); los casquillos y sobresellos; recubrimiento interno del cierre; sellos internos; puertos de administración; envolturas; accesorios de administración; y etiquetas.
Compuestos relacionados: Sustancias relacionadas estructuralmente con el principio activo, dichas sustancias pueden ser: impurezas y/o productos de degradación resultantes del proceso de producción o almacenamiento.
Datos de soporte: Datos, diferentes a los presentados en los estudios de estabilidad formales, que respaldan los procedimientos analíticos, el período de re análisis o vida útil propuesto y las condiciones de almacenamiento establecidas en la etiqueta. Tales datos incluyen: (1) datos de estabilidad en los primeros lotes de ruta sintética del IFA, lotes a pequeña escala de materiales, formulaciones de investigación no propuestas para comercialización, formulaciones relacionadas y producto presentado en envases y cierres distintos a los propuestos para la comercialización;(2) información sobre los resultados de las pruebas en envases y (3) otras razones científicas.
Diseño de extremos (bracketing): Es el diseño de un programa de estabilidad en el que solo las muestras de los extremos se evalúan en todos los tiempos del diseño completo. El diseño asume que la estabilidad de cualquier nivel intermedio está representada por la estabilidad de los extremos evaluados. Este diseño puede aplicarse a diferentes tamaños de envase o llenado en el mismo sistema de cierre de envase.
Diseño de matrices (matrixing): Es el diseño de un programa de estabilidad, tal que un subconjunto seleccionado del número total de las posibles muestras para todas las combinaciones de factores, se prueban en un punto de tiempo especificado. En el punto de tiempo siguiente, se ensaya otro subconjunto de muestras para todas las combinaciones de factores. El diseño asume que la estabilidad de cada subconjunto de muestras probadas representa la estabilidad de todas las muestras en un punto de tiempo dado. Las diferencias en las muestras para el mismo fármaco deben identificarse como, por ejemplo, cubriendo diferentes lotes, diferentes tamaños del mismo sistema de cierre de envases y, posiblemente, en algunos casos, diferentes sistemas de cierre de envases.
Empaque: Recipiente definitivo de distribución y comercialización dentro del que se coloca el envase primario que contiene el medicamento en su forma farmacéutica.
Envase: Recipiente que contiene un compuesto intermedio, ingrediente farmacéutico activo, excipiente o forma farmacéutica, y que está o puede estar en contacto directo con estos.
Envase impermeable: Tipo de envase que protege el contenido de la contaminación por líquidos, sólidos o vapores extraños; de la pérdida, eflorescencia, delicuescencia o evaporación en condiciones habituales o usuales de manejo, distribución, almacenamiento y distribución.
Envase primario: Recipiente que está en contacto directo con el producto en evaluación, por ejemplo, IFA, intermedio, PFT, etc.
Envase resistente a la luz: Tipo de envase que protege el contenido de los efectos de la luz, por medio de las propiedades específicas del material que lo compone, incluyendo los recubrimientos aplicados sobre los mismos.
Envase semipermeable: Recipiente que permite el paso de un solvente, frecuentemente el agua, pero previene el paso de la sustancia disuelta o soluto, resultando así en un incremento de la concentración a través del tiempo. Un ejemplo de envase semipermeable es el envase plástico de polietileno de baja densidad (PEBD).
Especificación: Corresponde a la combinación de pruebas y criterios de aceptación físicos, químicos, biológicos y microbiológicos, que determinan la conformidad para la utilización de un IFA o un PFT y sus componentes.
Especificación de liberación: Corresponde a la combinación de pruebas y de criterios de aceptación físicos, químicos, biológicos y microbiológicos, que determinan la conformidad de un IFA o un PFT al momento de su liberación. Por lo general las especificaciones del ensayo de valoración para liberación deben ser más estrechas que las especificaciones establecidas durante la vida útil y debe tener en cuenta el porcentaje de degradación obtenido en los estudios de estabilidad acelerados y naturales.
Especificación durante la vida útil: Corresponde a la combinación de pruebas y criterios de aceptación físicos, químicos, biológicos y microbiológicos, que determinan la conformidad hasta el periodo de re análisis o vida útil para un IFA o un PFT.
Estudios de estabilidad acelerados (Estudios a corto plazo): Son estudios diseñados para incrementar la velocidad de la degradación química y los cambios físicos de un IFA o de un PFT, empleando condiciones extremas de almacenamiento, como parte del programa de evaluación de estabilidad.
Estudios de estabilidad natural (Estudios a largo plazo): Son estudios diseñados para evaluar las características físicas, químicas, biológicas, y microbiológicas de un IFA o PFT, durante el tiempo de conservación y el periodo de almacenamiento previstos.
Estudios de estabilidad "On-Going": Luego de que se ha autorizado la comercialización del PFT, la estabilidad del mismo debe ser verificada de acuerdo con un programa de estabilidad continuo, que permita monitorear el producto durante el tiempo de vida útil y determinar que este permanece y puede permanecer dentro de especificaciones bajo las condiciones de almacenamiento establecidas en el Registro Sanitario. Estos estudios se programan por lo menos para un lote de producción por año, a menos que no se fabrique ninguno en el año.
Extractables: Materiales o componentes provenientes del recipiente y/o cierre, los que son transferidos dentro del contenido del IFA o del PFT a lo largo del tiempo.
Extrapolación: es la práctica de utilizar un conjunto de datos conocidos para inferir información sobre datos futuros.
Fecha de expiración (fecha de vencimiento/ fecha de caducidad): Fecha que señala el tiempo del cual se espera que el IFA o el PFT permanezcan dentro de especificaciones si es almacenado bajo las condiciones establecidas. Ningún IFA o PFT podrá ser utilizado más allá de la finalización de su vida útil estimada.
Fecha de re análisis: Fecha después de la cual se debe volver a analizar un IFA para asegurar que cumple con las especificaciones y puede ser utilizado en la manufactura del PFT.
Forma farmacéutica (Forma de dosificación): Combinación de IFA(s) y/o excipientes en cantidades y formas físicas diseñadas para permitir una administración exacta y eficiente del PFT al paciente humano o animal (p. ej. tableta, cápsula, solución, crema).
Humedad relativa: Contenido de vapor de agua existente en la atmósfera, expresada como el grado de saturación existente en el ambiente a unas condiciones de temperatura y presión atmosférica definidas.
Impureza: Cualquier componente de un IFA o un PFT, diferente a la entidad química definida como principio activo. Las impurezas pueden ser: Orgánicas (Compuestos relacionados, intermediarios de síntesis, productos de degradación), Inorgánicas (Sales inorgánicas, metales pesados) y Solventes residuales.
Ingrediente Farmacéutico Activo (IFA): Cualquier sustancia o mezcla de sustancias a ser usadas en la fabricación de forma farmacéutica y que, cuando se usa, se convierte en el ingrediente activo del PFT. Estas sustancias se emplean en el diagnóstico, cura, mitigación, tratamiento o prevención de la enfermedad o para afectar la estructura y función del cuerpo.
Intervalo de confianza: Corresponde a un rango de valores, cuya distribución es normal y en el cual se encuentra, con alta probabilidad, el valor real de una determinada variable. Esta "alta probabilidad" se ha establecido por consenso en 95%. Así, un intervalo de confianza de 95% nos indica que dentro del rango dado se encuentra el valor real de un parámetro con 95% de certeza.
Límites de confianza: Están constituidos por el menor y el mayor valor de un intervalo de confianza.
Límite inferior del 95% de confianza: Se denomina así al intervalo existente entre el valor medio correspondiente a un parámetro y el menor valor existente dentro del 95% más probable. Este límite suele ser empleado como el más adecuado para seleccionar un valor determinado, en atención a que presenta una probabilidad del 95% de ocurrencia o éxito.
Liposomas: Atributo para preparaciones de lípidos anfifílicos de hidrosolubilidad baja. Son medicamentos singulares con propiedades únicas que pueden estar en forma de solución o suspensión.
Lixiviable: Son entidades químicas, orgánicas o inorgánicas, que migran de los componentes del envase farmacéuticos a la formulación de medicamento.
Lote piloto (también denominado piloto industrial): Un lote de un IFA o de un PFT, elaborado en escala menor a la industrial, fabricado mediante un procedimiento totalmente representativo y que simula el proceso que va a ser aplicado a un lote manufacturado a escala completa (industrial). Por ejemplo, un lote a escala piloto es por lo general el que tiene un tamaño mínimo de 1/10 con relación al de escala de producción industrial o lo que sea mayor.
Nota: En situaciones excepcionales que serán establecidas por el Invima, y en casos particulares plenamente justificados por la empresa, se aceptarán tamaños de lote piloto inferior al aquí definido.
Lote de producción (Industrial): Es un lote de un IFA o de un PFT elaborado en escala de producción industrial, utilizando los equipos y las instalaciones de manufactura que se especificaron en la solicitud.
Lote primario: Es un lote de un IFA o un PFT utilizado en un estudio de estabilidad formal, del cual los datos de estabilidad son sometidos en una solicitud de Registro para propósitos de establecer un periodo de re análisis o un periodo de vida útil. Un lote primario de un IFA debe ser al menos un lote a escala piloto. Para un PFT, en el caso de estudios sobre tres lotes, dos de tres lotes deben ser al menos escala piloto y el tercer lote puede ser más pequeño si este es representativo con respecto a las etapas de manufactura críticas. Sin embargo, un lote primario también puede ser un lote de producción o industrial.
Muestra: Cantidad de unidades o partes de un todo, extraída con criterio racional y al azar, para asegurar que la misma es estadísticamente representativa del material a analizar. Las unidades deben ser evaluadas en forma independiente.
Muestra compuesta: En diseño experimental se refiere a la muestra conformada por más de una unidad de dosificación, la que debe ser procesada en forma conjunta, dando lugar a un solo dato que se analiza de forma individual.
Muestra simple: En diseño experimental se refiere a la muestra conformada por una sola unidad de dosificación, la que debe ser procesada en forma independiente, dando lugar a un solo dato que se analiza de forma individual.
Periodo de re análisis: Espacio de tiempo durante el cual el IFA o un excipiente, se espera que se mantenga dentro de sus especificaciones y por consiguiente puede ser utilizado en la manufactura de medicamentos, demostrando que ha sido almacenado bajo las condiciones definidas.
Producto a granel: Estado intermedio de un producto que ha completado todas las etapas del procesamiento, hasta el envasado final, pero sin incluir este último.
Producto de degradación (producto de descomposición): Es aquel o aquellos compuestos químicos que se generan como resultado de la inestabilización de la(s) molécula(s) original(es). Para los propósitos de los ensayos de estabilidad de los medicamentos descritos en esta guía, podría generarse como resultado del proceso de almacenamiento (p. ej.: hidrólisis, desamidación, oxidación, agregación, proteólisis, etc.).
Producto para reconstituir: Aquel PFT que requiere de un proceso adicional (p.ej. reconstitución o dilución) para su uso o administración final, en la forma indicada en las instrucciones de preparación.
Producto Farmacéutico Terminado (PFT): Producto que ha sido sometido a todas las etapas de producción, incluyendo el envasado en el recipiente final y el etiquetado.
Protocolo de estudio de estabilidad: Documento que describe con claridad el procedimiento a seguir para desarrollar un estudio de estabilidad definido. En él se establece tanto el diseño experimental, como la metodología para evaluar la información y obtener las conclusiones.
Pruebas de estabilidad: Serie de ensayos diseñados para obtener información sobre la estabilidad de un PFT, producto intermedio, IFA, etc., con el objetivo de definir su tiempo de vida útil y/o período de re análisis, bajo condiciones específicas de almacenamiento y envase.
Pruebas de estrés (del IFA): Estudios realizados para elucidar la estabilidad intrínseca del IFA. Estas pruebas forman parte de las estrategias de desarrollo y se lleva a cabo normalmente en condiciones más severas que las utilizadas para los estudios de estabilidad acelerados.
Pruebas de estrés (del PFT): Estudios realizados para evaluar el efecto de condiciones severas en el PFT, dichos estudios incluyen pruebas de foto estabilidad y pruebas específicas para algunos productos (Por ejemplo, inhaladores de dosis medidas, cremas, emulsiones, etc.).
Registro del estudio de estabilidad: Es el documento propiedad del titular, donde se lleva el registro de los resultados y cálculos de todas las pruebas y análisis efectuados, así como también las observaciones realizadas en los estudios de estabilidad. Este documento o archivo (con existencia en cualquier medio comprobable) debe estar refrendado por la persona responsable de la realización del estudio. Podrá ser sometido a inspección por la autoridad sanitaria pertinente.
Réplica: Datos obtenidos cuando una misma muestra es sometida dos o más veces a un mismo análisis o a determinaciones repetidas. Los resultados de las réplicas no pueden ser tomados como resultados de análisis de muestras independientes.
Reproceso: Reelaboración de todo o parte de un lote de un producto, de calidad inaceptable en una etapa definida de la producción, de tal forma que por medio de una o más operaciones adicionales, su calidad se modifica hasta ser aceptable según sus especificaciones.
Resultado atípico: Cualquier valor que esté dentro de las especificaciones establecidas, pero que es "irregular" o "diferente" de los demás valores de un mismo grupo de datos o se encuentra fuera de las tendencias de los resultados dentro del estudio de estabilidad.
Sistemas de envase y cierre: Se refiere al conjunto de los componentes de los envases que contienen y protegen la forma de dosificación, esto incluye los componentes de envase primario y secundario, si este último está diseñado para proporcionar una protección adicional.
Spray: Es una forma farmacéutica que contiene fármacos en estado líquido, como una solución o como una suspensión, y está destinado para su administración como una nube de gotitas. Los sprays se diferencian de los aerosoles en que los atomizadores no están presurizados.
Tiempo de vida útil (Periodo de vida útil o vida de anaquel): Es el periodo de tiempo durante el cual un IFA o un PFT, si es almacenado bajo las condiciones establecidas, se espera que cumpla con las especificaciones. El tiempo de vida útil es utilizado para establecer la fecha de expiración de cada lote.
Tiempo de vida útil definitivo: Es el tiempo de vida útil finalmente asignado a un producto y determinado por el desarrollo de los estudios de estabilidad natural, realizados sobre el producto en el envase y empaque en que se va a comercializar.
Tiempo de vida útil tentativa (provisional): Período de vida útil determinado por proyección de los datos obtenidos en estudios de estabilidad acelerada y natural, aplicados al producto en el mismo envase primario en que va a ser comercializado.
Tolerancia en las condiciones de almacenamiento: Son las variaciones aceptables de temperatura y humedad relativa, en las instalaciones de almacenamiento para los estudios de estabilidad.
Validación: Procedimiento para establecer la evidencia documentada, que provee un alto grado de seguridad, de que un proceso específico entrega los resultados esperados.
9. APÉNDICE 1. Árbol de Decisión
Árbol de decisión para la evaluación de datos para la estimación del periodo de vida útil PFTs (excepto para los productos congelados).
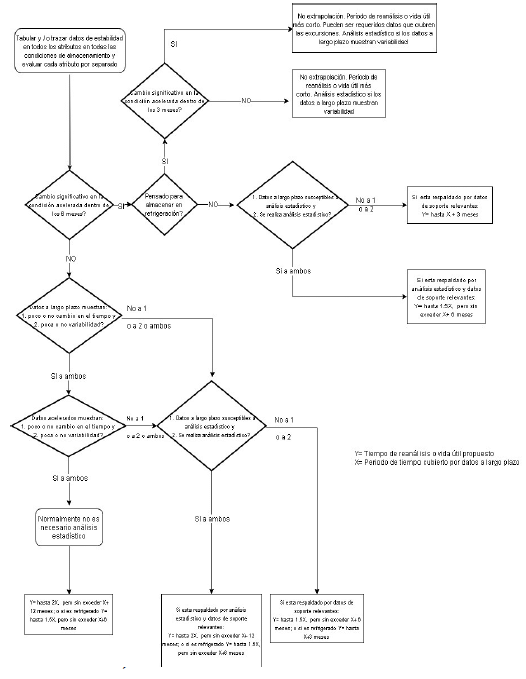
10. BIBLIOGRAFÍA
1. Anexo 2 Estudios de Estabilidad de ingredientes farmacéuticos activos y productos farmacéuticos terminados, Informe 43 OMS, 2009.
2. ICH Harmonised Tripartite Guideline.Stability Testing of New Drug Substances and Products. ICHQ1A(R2), February 2003.
3. ICH Harmonized Tripartite Guideline Stability Testing Photostability Testing of New Drug Substances and Products Q1B, November 1996.
4. ICH Harmonized Tripartite Guideline Bracketing and Matrixing Designs for Stabilitybility Testing of New Drug Substances and Products Q1D, February 2002.
5. Guía para el Desarrollo y Presentación de los Estudios de Estabilidad de Medicamentos; mayo de 1995, Resolución 2514 de 1995.
6. USP, Farmacopea de los Estados Unidos de América.
7. ICH Harmonized Tripartite Guideline Validation of Analytical Procedures: Text and Methodology Q2 (R1), November 2005.
8. Manual de Normas Técnicas de Calidad. Guía Técnica de Análisis. Ministerio de Salud, INVIMA. Tercera Revisión. 2002.
9. ICH Harmonised Tripartite Guideline. Evaluation for Stability Data Q1E, February 2003.
10. Huynh-Ba, K. Handbook of Stability Testing in Pharmaceutical Development Regulations, Methodologies and Best Practices. Springer 2009.
11. NirmalK, Ajeya J. Temperature excursión management: A novel approach of quality system in pharmaceutical industry. Saudi Pharmaceutical Journal (2017) 25, 176– 183.
12. Ammann C.Stability Studies Needed to Define the Handling and Transport Conditions of Sensitive Pharmaceutical or Biotechnological Products. AAPS Pharm Sci Tech, Vol. 12, número 4, December 2011.
13. Seevers R, Bishara R, Harber PJ Lucas T. Designing stability studies for time/ temperature exposure. American Pharmaceutical Outsourcing. 2005; 6(5): 18, 20, 21, 23, 55.
14. Lucas T, Bishara R, Seevers R. A stability program for the distribution of drug products. pp. 68–73, s.l.: Pharmaceutical Technology, July 2004.
Las notas de vigencia, concordancias, notas del editor, forma de presentación y disposición de la compilación están protegidas por las normas sobre derecho de autor. En relación con estos valores jurídicos agregados, se encuentra prohibido por la normativa vigente su aprovechamiento en publicaciones similares y con fines comerciales, incluidas -pero no únicamente- la copia, adaptación, transformación, reproducción, utilización y divulgación masiva, así como todo otro uso prohibido expresamente por la normativa sobre derechos de autor, que sea contrario a la normativa sobre promoción de la competencia o que requiera autorización expresa y escrita de los autores y/o de los titulares de los derechos de autor. En caso de duda o solicitud de autorización puede comunicarse al teléfono 617-0729 en Bogotá, extensión 101. El ingreso a la página supone la aceptación sobre las normas de uso de la información aquí contenida.