|
|
|
RESOLUCIÓN 184 DE 2024
(febrero 8)
Diario Oficial No. 52.663 de 8 de febrero de 2024
MINISTERIO DE SALUD Y PROTECCIÓN SOCIAL
Por la cual se adopta la Política de Dispositivos Médicos.
EL MINISTRO DE SALUD Y PROTECCIÓN SOCIAL,
en uso de sus facultades legales, en especial las conferidas en el artículo 86 de la Ley 1438 de 2011, el artículo 23 de la Ley 1751 de 2015, el numeral 9 del artículo 2o y el numeral 1 del artículo 25 ambos del Decreto número 4107 de 2011 y
CONSIDERANDO:
Que el artículo 49 de la Constitución Política de 1991 determina una garantía universal de accesibilidad a los servicios de promoción, protección y recuperación de la salud y señala que corresponde al Estado organizar, dirigir y reglamentar la prestación de servicios de salud a los habitantes, teniendo en cuenta los principios de eficiencia, universalidad y solidaridad.
Que la 60° Asamblea Mundial de la Salud adoptó la Resolución WHA60.29 en mayo de 2007, en la cual, basados en la necesidad de ampliar los conocimientos especializados sobre tecnologías sanitarias, sobre todo en los dispositivos médicos, instó a los países miembros a jerarquizar las necesidades y los recursos basados en información, implantar sistemas de evaluación, planificación, adquisición y gestión de tecnologías y a elaborar directrices sobre prácticas adecuadas de fabricación y reglamentación.
Que en el artículo 2o de la Ley 1751 de 2015 estatutaria del derecho a la salud establece que el derecho fundamental a la salud comprende el acceso a los servicios de salud de manera oportuna, eficaz y con calidad para la preservación, el mejoramiento y la promoción de la salud e indica adicionalmente que el Estado adoptará políticas para asegurar la igualdad de trato y oportunidades en el acceso a las actividades de promoción, prevención, diagnóstico, tratamiento, rehabilitación y paliación para todas las personas.
Que en la precitada ley, el literal j) del artículo 5o, en lo referente a las obligaciones del Estado, determina que este debe intervenir el mercado de medicamentos, dispositivos médicos e insumos en salud con el fin de optimizar su utilización, evitar las inequidades en el acceso, asegurar la calidad de los mismos o en general cuando pueda derivarse una grave afectación de la prestación del servicio; así mismo su artículo 10, enlista los derechos relacionados con la prestación del servicio de salud que tienen las personas, y señala en su literal i) que estos tienen derecho a la provisión y acceso oportuno a las tecnologías y a los medicamentos requeridos.
Que el Decreto número 4107 de 2011, el cual determina los objetivos y la estructura del Ministerio de Salud y Protección Social, establece en el numeral 9 del artículo 2o que le corresponde a esta Entidad formular, adoptar y evaluar la política de dispositivos médicos, así como, establecer y desarrollar mecanismos y estrategias dirigidas a optimizar la utilización de los mismos.
Que, de acuerdo con el contexto internacional, los reactivos de diagnóstico in vitro se encuentran incluidos dentro de los dispositivos médicos, por lo que en esta política se entenderá que los dispositivos médicos, comprenden también a los reactivos de diagnóstico in vitro, esto en concordancia con el Decreto número 3770 de 2004 que reglamenta el régimen de registros sanitarios y la vigilancia sanitaria de los reactivos de diagnóstico in vitro para exámenes de especímenes de origen humano y el Decreto número 4725 de 2005 norma que contiene las disposiciones que autorizan la comercialización de los dispositivos médicos en Colombia.
Que en cumplimiento a lo dispuesto en el artículo 8o de la Ley 1437 de 2011 y el Anexo 1 del Decreto número 1081 de 2015, el presente proyecto de resolución fue publicado para comentarios de la ciudadanía y grupos de interés, en la página web del Ministerio, durante el periodo comprendido entre el día 28 de agosto de 2023 y el día 15 de septiembre de 2023.
Que en virtud de lo anterior, se hace necesario expedir la Política Nacional de Dispositivos Médicos la cual surge como resultado de un exhaustivo diagnóstico realizado en el sector salud por parte de este Ministerio y la cual contiene el conjunto de acciones que permitirán mejorar el acceso equitativo a los dispositivos médicos en Colombia, optimizar su utilización y contribuir al fortalecimiento del Sistema General de Seguridad Social en Salud (SGSSS) por medio de la integración de todos los factores que inciden en la disponibilidad, calidad, desempeño, continuidad, uso seguro y adecuado de estas tecnologías en salud.
En mérito de lo expuesto,
RESUELVE:
ARTÍCULO 1o. La presente resolución tiene como objeto adoptar la Política de Dispositivos Médicos contenida en el Anexo Técnico, el cual hace parte integral del presente acto administrativo.
ARTÍCULO 2o. La Dirección de Medicamentos y Tecnologías en Salud con el apoyo de la Oficina Asesora de Planeación y Estudios Sectoriales del Ministerio de Salud y Protección Social realizarán un seguimiento anual a la ejecución de las acciones propuestas para el cumplimiento del objetivo de la presente política, presentando informes de avance anuales en el periodo comprendido entre 2024 y 2026 y un informe de cierre final en el año 2027.
ARTÍCULO 3o. La presente resolución rige a partir de la fecha de su publicación.
Publíquese y cúmplase.
Dada en Bogotá, D. C., a 8 de febrero de 2024.
El Ministro de Salud y Protección Social,
Guillermo Alfonso Jaramillo Martínez.
POLÍTICA DE DISPOSITIVOS MÉDICOS.
Tabla de contenido
INTRODUCCIÓN
PRINCIPIOS
ANTECEDENTES Y JUSTIFICACIÓN
MARCO CONCEPTUAL
Generalidades de los DM en Colombia
Disponibilidad y asequibilidad
Rectoría y gobernanza
Eficacia y Efectividad
Sostenibilidad
Sistemas de información
Ciclo de vida
Investigación y desarrollo e Innovación
Evaluación de tecnologías en salud (ETES)
Regulación de precios
Calidad y seguridad
Vigilancia Posmercado
Gestión de Tecnologías
DIAGNÓSTICO
Contexto nacional de los DM.
Problema identificado
Causas
1. Carencia de un sistema de información de DM que integre las herramientas existentes e identifique las que se requieran para responder a las necesidades de toma de decisiones en el sector.
2. Insuficientes lineamientos que contribuyan al fortalecimiento de la investigación, el desarrollo y la práctica clínica en materia de DM.
3. Debilidades en la rectoría y gobernanza sobre los DM con escazas capacidades técnicas en los actores.
4. Escasas estrategias para fortalecer la disponibilidad y la asequibilidad de DM priorizados, que respondan al perfil epidemiológico de la población colombiana.
5. Débiles lineamientos posmercado, regulación y reglamentación técnica que hagan énfasis en la promoción de la calidad y seguridad de los DM.
6. Escasos lineamientos para la gestión, así como para el uso adecuado de DM.
OBJETIVOS
Objetivo general
Objetivos específicos
PLAN DE ACCIÓN
SEGUIMIENTO
FINANCIAMIENTO
BIBLIOGRAFÍA
SIGLAS Y ABREVIATURAS
ASIS. Análisis de la Situación de Salud
CEPAL. Comisión Económica para América Latina y el Caribe
CNPMDM. Comisión Nacional de Precios de Medicamentos y Dispositivos Médicos
COPNIA. Consejo Profesional Nacional de Ingeniería
DANE. Departamento Administrativo Nacional de Estadística
DM. Dispositivo médico
DMTS. Dirección de Medicamentos y Tecnologías en Salud
EPP. Elementos de protección personal
ETES. Evaluación de tecnologías en salud
GTS. Gestión de tecnologías en salud
IETS. Instituto Nacional de Tecnologías en Salud
IMDRF. Foro Internacional de Reguladores de Dispositivos Médicos
INVIMA. Instituto Nacional de Vigilancia de Medicamentos y Alimentos
IVC. Inspección, vigilancia y control
MSPS. Ministerio de Salud y Protección Social
OMS. Organización Mundial de la Salud
OPS. Organización Panamericana de la Salud
PC. Permisos de Comercialización
RDIV. Reactivos de diagnóstico in vitro
RS. Registros Sanitarios
SGSSS. Sistema General de Seguridad Social en Salud
SICA. Sistema de la Integración Centroamericana
SISDIS. Sistema de Información de Precios de Dispositivos Médicos
SISPRO. Sistema Integrado de Información para la Protección Social
TIC. Tecnologías de la Información y las Comunicaciones
INTRODUCCIÓN
Los dispositivos médicos (DM) son un componente de la esencia de la prestación de servicios de salud en tanto que su acceso hace parte de la materialización del derecho a la salud. Esta definición, a nivel internacional, también comprende a los reactivos de diagnóstico in vitro (RDIV), razón por la cual cuando se mencione DM se entenderá incluidos los RDIV.
Los DM representan la respuesta tecnológica de alta utilización en los sistemas de salud implicándole un alto costo, dado a que estos participan en las fases de promoción, prevención, tratamiento, rehabilitación y paliación de la atención en salud.
Según la Organización Mundial de la Salud (OMS), para 2017 se estimaba que había alrededor de 2 millones de tipos diferentes de DM en el mercado mundial, clasificados en más de 22.000 grupos de DM genéricos (OMS, 2017), lo que evidencia la presencia de DM en el mercado y su importancia en la atención en salud. En ese mismo año, la OMS insta a los Estados Miembros a que formulen “estrategias y planes nacionales adecuados para la evaluación y gestión de los dispositivos médicos” (OMS, 2012).
Por su parte, Colombia desde el año 2011, dispuso mediante la Ley 1438 la formulación de la Política de Dispositivos Médicos, por lo que, siguiendo los posteriores lineamientos de la OMS, el Ministerio de Salud y Protección Social (MSPS) ha venido trabajando en esta etapa de formulación, concretamente presentando un exhaustivo diagnóstico en el año 2019 y una propuesta de Política de Dispositivos Médicos en el año 2022.
De esta forma, este documento presenta la Política de Dispositivos Médicos para Colombia en el horizonte temporal 2023-2026, con las acciones sectoriales que permitan al MSPS y sus entidades adscritas establecer las estrategias para fortalecer a corto plazo el sector de DM.
El problema central identificado en esta política se enmarca en las dificultades en el acceso equitativo de la población colombiana a los DM y desintegración de los factores que inciden en la disponibilidad, calidad, continuidad, uso seguro y adecuado en la salud individual y colectiva para contribuir con los resultados en salud que se dan en el Sistema General de Seguridad Social en Salud.
Las causas de este problema se han clasificado en seis grandes categorías: (i) carencia de un sistema de información que integre las herramientas existentes e identifique las que se requieran para responder a las necesidades de toma de decisiones en el sector, (ii) insuficientes lineamientos que contribuyan al fortalecimiento de la investigación, el desarrollo y la práctica clínica en materia de DM, (iii) debilidades en la rectoría y gobernanza sobre los DM con escazas capacidades técnicas en los actores, (iv) escasas estrategias para fortalecer la disponibilidad y la asequibilidad de DM priorizados, que respondan al perfil epidemiológico de la población colombiana, (v) débiles lineamientos posmercado, regulación y reglamentación técnica que hagan énfasis en la promoción de la calidad y seguridad de los DM y (vi) escasos lineamientos para la gestión, así como para el uso adecuado de DM.
De esta forma se definen las estrategias a desarrollar en el siguiente trienio, en términos de gobernanza, acceso equitativo y asequible y sostenible, sistemas de información y los avances sugeridos para investigación y desarrollo, así como el fortalecimiento de la industria local, todo esto, orientado al logro de acceso a los DM, que permita la satisfacción de las necesidades de la salud pública de la población colombiana.
PRINCIPIOS
La política de dispositivos médicos contempla los siguientes principios:
Accesibilidad. Los DM deben ser accesibles a todos, en condiciones de igualdad, dentro del respeto a las especificidades de los diversos grupos vulnerables y al pluralismo cultural. La accesibilidad comprende la no discriminación, la accesibilidad física, la asequibilidad económica y el acceso a la información.
Asequibilidad. Medida en que los usuarios a quienes está destinado un DM pueden pagarlo.
Equidad. Los DM deben estar disponibles para toda la población independientemente de su capacidad de pago y condiciones particulares, evitando que prestaciones individuales no pertinentes de acuerdo con criterios técnicos y científicos pongan en riesgo los recursos necesarios para la atención del resto de la población.
Integralidad. La atención en salud de la población debe incluir los DM necesarios para la promoción, prevención, diagnóstico, tratamiento, rehabilitación y paliación de la enfermedad.
Calidad. Los DM deberán estar centrados en el usuario, ser apropiados desde el punto de vista médico y técnico y responder a estándares de calidad aceptados por las comunidades científicas. Ello requiere, entre otros, personal de la salud adecuadamente competente, enriquecida con educación continua e investigación científica y una evaluación oportuna de la calidad de los servicios y tecnologías ofrecidos.
Eficiencia. Es la óptima relación entre los recursos disponibles para obtener los mejores resultados en salud y calidad de vida de la población.
Disponibilidad. Es la presencia de un DM en el mercado colombiano.
Oportunidad. Los DM deben proveerse sin dilaciones.
Sostenibilidad. Los DM que se enmarcan en las prestaciones que reconoce el sistema se financiarán con los recursos destinados por la ley para tal fin, los cuales deberán tener un flujo ágil y expedito. Las decisiones que se adopten en el marco del Sistema General de Seguridad Social en Salud deben consultar criterios de sostenibilidad fiscal. La administración de los fondos del sistema no podrá afectar su flujo de recursos.
ANTECEDENTES Y JUSTIFICACIÓN
La Política de Dispositivos Médicos ha sido referenciada desde el artículo 86 de la Ley 1438 de 2011 y reiterada en la Ley 1751 de 2015 Ley estatutaria del derecho a la salud. En ese periodo de tiempo entró a funcionar la Dirección de Medicamentos y Tecnologías en Salud (DMTS) para fortalecer la estructura técnica, tanto de talento humano como financiero, que permitieron adelantar estudios que sirvieron de insumos para su construcción.
En este panorama, aunado con el fortalecimiento gradual y la apropiación del presupuesto de los últimos años, se logró contar con un diagnóstico del sector de los DM, elaborado por el Instituto Nacional de Tecnologías en Salud (IETS) en 2019, lo que permitió en el año 2022 socializar y validar con los actores, los avances de la construcción del presente documento.
Por otra parte, el crecimiento exponencial que han tenido los DM en el mundo confirma la rapidez de los avances tecnológicos y el aumento en su uso, generando la necesidad de afianzar el concepto de calidad en los servicios de salud, de revisar el contexto de uso de los DM y la posibilidad de que ocurra un incremento en los costos de la atención en salud.
Otros factores reconocidos a nivel global como problemáticos relacionados a los DM, incluye la débil gestión, carencia de mecanismos de selección y priorización, bajos recursos para la adquisición, falta de información y capacitación del talento humano y una evidente discordancia entre la regulación, las medidas de control y el mercado (OMS, 2012).
Como respuesta a dichas problemáticas, la Asamblea Mundial de la Salud en mayo de 2007, adoptó la Resolución WHA60.29, en la cual se plasmó la necesidad de establecer prioridades en la selección que cubran la carga de enfermedad, ampliar los conocimientos sobre tecnologías sanitarias, especialmente DM, garantía de la disponibilidad y a precios asequibles con el objetivo de “asegurar la mejora del acceso, la calidad y el uso de productos médicos y tecnologías sanitarias” (OMS, 2007).
De igual forma también instó a los Estados Miembros a que en sus agendas incluyan la formulación de “estrategias y planes nacionales para la implantación de sistemas de evaluación, planificación, adquisición y gestión de tecnologías sanitarias, en particular dispositivos médicos, en colaboración con personal dedicado a la evaluación de las tecnologías sanitarias y la ingeniería biomédica” (OMS, 2017, p. 2), y a que “elaboren directrices nacionales o regionales sobre prácticas adecuadas de fabricación y reglamentación, instituyan sistemas de vigilancia y otras medidas para garantizar la calidad, seguridad y eficacia de los dispositivos médicos y, cuando corresponda, participen en la armonización internacional” (OMS, 2017, p. 2). Colombia como país miembro de la OMS, viene adoptando lineamientos desde hace más de una década sobre DM, de igual modo, ha desarrollado normatividad local bajo la legislación general como la Ley 09 de 1979 que en su título 11, establece la vigilancia y el control de productos que incidan en la salud pública. Igualmente, para el año 1986 el Decreto número 2092, en los artículos 92 y 93, determinó que las suturas y algunos materiales de curación requerían registro sanitario.
En el año 1993, se promulgó la Ley 100 que consagró la equidad y la obligatoriedad como fundamentos del servicio público de la seguridad social en salud. En esta ley se incluyeron algunos lineamientos relacionados con DM tales como: la creación del Instituto Nacional de Vigilancia de Medicamentos y Alimentos (Invima) en el artículo 245 y un paquete de servicios de salud que dispuso estas tecnologías de salud a toda la población afiliada.
Posteriormente la Ley 1122 de 2007, que modifica la Ley 100 de 1993, en el literal d) del artículo 34, introdujo mecanismos y establecimientos para la distribución y venta de DM; por su lado la Ley 1438 de 2011, incluyó el control de precios de los DM, en la hoy Comisión Nacional de Precios de Medicamentos y Dispositivos Médicos (CNPMDM), estableció además la codificación de DM ratificada en el artículo 117 del Decreto Ley 019 de 2012 y ordenó la definición de la política farmacéutica, de insumos y DM a nivel nacional, la cual tendría como objetivos garantizar acceso, la calidad y el uso racional de estas tecnologías en salud a nivel nacional.
De igual forma, la Ley Estatutaria 1751 de 2015 que representa uno de los avances legislativos de mayor relevancia en el país, en términos de reconocimiento de derechos para la población, al considerar la salud como derecho fundamental, desarrolla en el capítulo IV lineamientos enfocados a la regulación sanitaria de medicamentos, DM e insumos para la salud, innovación, ciencia y tecnologías en salud. Así mismo, destacan la renovación de la política farmacéutica nacional, programática e integral y en la que se identifiquen las estrategias, prioridades, mecanismos de financiación, adquisición, almacenamiento, producción, compra y distribución de los insumos, tecnologías y medicamentos, basadas en criterios de necesidad, calidad, costo efectividad, suficiencia y oportunidad.
Ahora bien, el MSPS inicia la regulación sanitaria específica para DM, cuando en 2004 expide el Decreto número 3770, que establece las disposiciones del régimen de registro y vigilancia sanitaria de los reactivos de diagnóstico in vitro (RDIV), al año siguiente expide el Decreto número 4725 de 2005 considerado norma marco, dado su alcance al universo de los DM, cuyo objeto igualmente es el registro sanitario y la vigilancia sanitaria, con el propósito de garantizar la efectividad, seguridad y calidad de los productos, para la protección de la salud pública de la población.
Cabe mencionar así mismo, la existencia de reglamentación mediante resoluciones de los referidos decretos y de los DM sobre medida, adicional a las modificaciones que se han tenido en los últimos años.
A partir de 2013, los reglamentos relacionados a DM y otras tecnologías en salud, se diseñan bajo los lineamientos del modelo de inspección, vigilancia y control sanitario establecido en la Resolución número 1229, que define la obligatoriedad de implementar mecanismos que reduzcan las “amenazas y riesgos asociados a la producción e intercambio de bienes y servicios de uso y consumo humano”.
Con relación a control de precios de DM, la CNPMDM ha expedido las siguientes circulares: 01 de 2015, 02 de 2015, 02 de 2016, 02 de 2017, 05 de 2018, 13 de 2021 y 14 de 2022. Como resultado, actualmente se tienen dos mercados relevantes de DM endovasculares coronarios y anticonceptivos, en régimen de libertad vigilada (únicamente reporta el precio de comercialización); un DM, estent coronario medicado en control directo de precios (establece un precio máximo de venta). Para el reporte al Sistema Integrado de Información para la Protección Social (SISPRO) el MSPS cuenta con el Sistema de Información de Precios de Dispositivos Médicos (SISDIS).
Por otro lado, es importante mencionar el hito que marca la historia del mundo actual, al enfrentar la pandemia por COVID-19 en el año 2020, en donde Colombia tuvo que adoptar medidas para prevenir y controlar la propagación de la enfermedad y mitigar sus efectos. Para dar respuesta a la grave situación de salud generada por la pandemia, se definieron acciones que abrevien las condiciones de fabricación, adquisición, compra y gestión de tecnologías sanitarias, específicamente para DM, en su mayoría consumibles, elementos de protección personal (EPP), equipos biomédicos y reactivos de diagnóstico in vitro.
En este tema, según estudios de la Comisión Económica para América Latina y el Caribe (CEPAL) y del Sistema de la Integración Centroamericana (SICA), en la crisis provocada por el COVID-19 se identifica la fragilidad de la gestión y el papel de la gobernanza, la planificación y el gobierno digital que se vio reflejado en todos los componentes de la atención, y en especial en el abastecimiento de las tecnologías sanitarias (CEPAL, 2021).
En este contexto, en cuanto a DM es fundamental establecer una política integral, que contribuya al fortalecimiento del SGSSS, propender por la seguridad, calidad y desempeño de los DM, los cuales son tecnologías en salud cruciales para la prestación de la atención en salud y así poder responder a las necesidades, en términos de prevención y a lo largo del ciclo de atención: investigación, diagnóstico, tratamiento y manejo, seguimiento y rehabilitación.
MARCO CONCEPTUAL
A continuación, se tratarán los principales conceptos en torno a los DM en Colombia que son la base para el desarrollo de la Política de Dispositivos Médicos y que permitirán consolidar los términos y establecer el lenguaje común.
Generalidades de los DM en Colombia
Según la normatividad vigente en Colombia, las tecnologías sanitarias incluyen las actividades, procedimientos e intervenciones, medicamentos, dispositivos, productos, servicios y procedimientos usados en la prestación de servicios de salud, así como los sistemas organizativos y de soporte con los que se presta la atención en salud (Resolución número 2366 de 2023).
Dentro de las tecnologías en salud se encuentran los DM y los RDIV, aunque en Colombia su reglamentación específica es diferente, internacionalmente los RDIV son considerados DM, en atención a la definición establecida en el año 2018 por el Foro Internacional de Reguladores de Dispositivos Médicos (IMDRF por las siglas en inglés de International Medical Device Regulators Forum) y que agrupa los usos de los DM y RDIV. Para este documento, cuando se hable de DM se incluyen también los RDIV.
Los DM por su lado, constituyen un universo de productos que incluye desde un aplicador de algodón o una gasa, cuya manufactura no es compleja, hasta un ventilador mecánico o un tomógrafo. El Decreto número 4725 de 2005, define DM como cualquier instrumento, aparato, máquina, software, equipo biomédico u otro artículo que se usen en diagnóstico o tratamiento de una enfermedad, como soporte de una estructura anatómica o de un proceso fisiológico; también lo son todos aquellos que se usan en el control de la concepción, en el diagnóstico y cuidado del embarazo, en la atención del parto y recién nacidos y los usados como productos de desinfección o esterilización de los mismos DM.
Así mismo, el Decreto número 3770 de 2004 define el RDIV como “un producto reactivo, calibrador, elaborado en material de control, utilizado sólo o en asociación con otros, destinado por el fabricante a ser utilizado in vitro para el estudio de muestras procedentes del cuerpo humano (…)” (Decreto número 3770 de 2004).
Con estas definiciones se puede concluir que la diversidad es una característica inherente en el mundo de los DM. Ahora bien, los DM por su definición son usados en todas las etapas de la atención en salud y su uso seguro es relevante, ya que impacta directamente la vida o la salud de las personas.
Para su abordaje, la DMTS del MSPS ha realizado una clasificación en tipologías y subtipologías así:
1. Tipología consumible. Compuesta por cuatro subtipologías:
1.1 Subtipología consumibles o insumos. Conocidos como de único uso, fungibles, descartables o desechables (Minsalud, 2019).
1.2 Subtipología implantables. Dispositivos utilizados en diferentes especialidades quirúrgicas que son implantados de forma temporal o definitiva en un paciente (Minsalud, 2019). Incluyen aquellos que son sobremedida implantables.
1.3 Subtipología reactivo In vitro y de diagnóstico in vitro. Sustancia o compuesto químico o biológico, que solo o en asociación es diseñado para análisis o pruebas de muestras de origen humano con el fin de proporcionar información relativa a la situación en estudio (Decreto número 3770 de 2004).
1.4 Subtipología reutilizables. Instrumento destinado a fines quirúrgicos para cortar, perforar, cerrar, escarificar, raspar, pinzar, retraer, recortar u otros procedimientos similares, sin estar conectado a ningún dispositivo médico activo y que puede volver a utilizarse una vez efectuados todos los procedimientos pertinentes (Decreto número 4725 de 2005).
2. Tipología equipos biomédicos.
Tecnologías operacionales y funcionales que reúnen sistemas y subsistemas eléctricos, electrónicos o hidráulicos, incluidos los programas informáticos que intervienen en su funcionamiento. (Minsalud, 2019).
3. Tipología dispositivos sobre medida.
Tecnologías personales elaboradas, fabricados, siguiendo una prescripción médica para un usuario particular, para suplir deficiencias visuales/oculares, auditivas, bucales y funcionales del sistema musculoesquelético (Minsalud, 2019).
Dispositivos médicos sobre medida de tecnología ortopédica: Suplen deficiencias del funcionamiento de miembros superiores, inferiores y otras estructuras músculo esqueléticas. (Minsalud, 2019).
Dispositivos médicos sobre medida de salud visual y ocular: Suplen deficiencias, ayudas ópticas y no ópticas para baja visión, tratamiento de problemas ortópticos, y pleópticos, estereogramas que requieran correspondencia dióptrica y filtros de lentes (Minsalud, 2019).
Dispositivos médicos sobre medida de ayuda auditiva: Suplen deficiencias auditivas (Minsalud, 2019).
Dispositivos médicos sobre medida bucales: Suplen las deficiencias dentales e integran la aparatología de ortodoncia, las prótesis dentales y las órtesis intrabucales (Minsalud, 2019).
4. Tipología DM como software.
Software destinado a ser utilizado para uno o más fines médicos que realizan estos propósitos sin ser parte de un dispositivo médico de hardware (FDA, 2023).
5. Tipología borderline.
Son aquellos dispositivos que por sus características intrínsecas no está claro desde el principio si está comprendido como dispositivo médico, medicamento, cosmético, o un producto de consumo general, por lo que se consideran productos frontera (borderline). (Borderline and Classification Working Group - Medical Device Coordination Group, 2022).
Disponibilidad y asequibilidad
Para que el SGSSS pueda brindar una atención efectiva e integral a la población colombiana se requiere de condiciones de disponibilidad de las tecnologías en el mercado local a precios asequibles que permitan el acceso seguro.
La OMS propone que los países establezcan acciones para mejorar el acceso en todo el mundo a unos DM idóneos, y que se propenda porque estos sean efectivos como medida terapéutica, que sean los necesarios para atender a la población y que cuenten con la calidad bajo estándares internacionales (OMS, 2012).
La accesibilidad o el acceso se refiere a “la capacidad de las personas de obtener tecnologías sanitarias de buena calidad cuando se necesitan y de utilizarlas adecuadamente” (OMS, 2012). La disponibilidad por su lado, “es la presencia de un DM en el mercado” (OMS, 2012). Finalmente, la asequibilidad está definida como la medida en que los usuarios a quienes está destinado un servicio de salud o producto sanitario pueden pagarlo (OMS, 2012).
En este sentido, la OMS promueve la utilización de DM prioritarios, adecuados al contexto o entorno al que está destinado; es decir, es preciso asociar el DM a la necesidad de salud, a fin de maximizar su efectividad y disminuir el gasto en DM inútiles, utilizando estudios y encuestas como el Análisis de la Situación de Salud (ASIS) y datos nacionales del Departamento Administrativo Nacional de Estadística (DANE) que permiten determinar los DM requeridos para manejar la carga de enfermedades existente en el país.
Los elementos de acceso, disponibilidad y asequibilidad son constitutivos de una adecuada gestión del ciclo de vida de los DM y contribuyen a una atención integral de la población.
Rectoría y gobernanza
Otros aspectos claves para la Política de DM es el abordaje a la rectoría y la gobernanza necesarias para establecer el rol regulatorio y el establecimiento de las reglas que deben regir en el desempeño de los actores y de los agentes del SGSSS para favorecer la atención integral, segura y con calidad, esto en concordancia, con la legislación vigente colombiana, los estándares internacionales y los lineamientos de la OMS.
Para ilustrar lo mencionado se puede considerar lo afirmado por Muthuri y Gatwiri que el alcance de la gobernanza en materia de salud va más allá del rol de rectoría, dado que incluye a parte de los acuerdos, los procesos, relaciones entre actores, reglas, mecanismos de control y rendición de cuentas (Kirigia J, 2011). Adicionalmente, la gobernanza también es el proceso mediante el cual los actores de una sociedad deciden sus objetivos de convivencia, fundamentales y coyunturales, y las formas de coordinarse para realizarlos: su sentido y su capacidad de dirección (Aguilar, 2006).
Por su parte, la rectoría según la OPS es una función indelegable e ineludible del Estado, ejercida a través de la autoridad de salud nacional, en Colombia el Ministerio de Salud y Protección Social (OPS y USAID, 2007). Esta rectoría debe contemplar dimensiones como: (i) conducción sectorial, (ii) regulación, (iii) financiamiento, (iv) aseguramiento, (v) armonización, (vi) ejecución de las funciones esenciales de salud pública. (Molina- Marín, 2012).
En este contexto, Colombia viene desarrollando la gobernanza y regulación incluidos en la reglamentación específica y a través de esta política que se convierte en el eje articulador.
Eficacia y Efectividad
Otro de los elementos a considerar en la Política de DM hace referencia a la eficacia y efectividad, la eficacia entendida como el logro del resultado esperado dentro de un entorno controlado, dado que según la definición (Monter, 2008) “es el grado en que una determinada intervención origina un resultado beneficioso en ciertas condiciones, medido en el contexto de un ensayo clínico controlado”; mientras la efectividad está relacionada al uso de los DM en términos del desempeño, así se encuentra definido por el IETS (IETS, 2014), como, “el beneficio (p. ej. en resultados en salud) que supone utilizar una tecnología para un determinado problema en condiciones generales o habituales” y definida por el IMDRF como la “capacidad de un dispositivo médico para proporcionar resultados clínicamente significativos de acuerdo con su uso previsto, conforme lo declara el fabricante” (traducido del inglés) (IMDRF,2019).
La eficacia y efectividad también están ligadas a los métodos, procedimientos, técnicas definidas en estándares internacionales para la fabricación y control de calidad, con el fin de generar DM aptos en el desempeño según su diseño, para que responda a la necesidad del paciente de manera segura.
Sostenibilidad
Para que el SGSSS pueda funcionar y cumplir con la misión de brindar salud integral y universal, bienestar a la población colombiana, así como para proporcionar los resultados en salud esperados, necesita del apalancamiento con recursos financieros permanentes y disponibles que le permita ser sostenible en el tiempo (Ley 1438 de 2011).
Al respecto, la OMS afirma “un sistema de salud sostenible como aquel que asegura el acceso equitativo a medicamentos esenciales, vacunas y tecnologías, mientras que recauda fondos adecuados para la salud, con el fin de garantizar que las personas puedan utilizar los servicios necesarios y estén protegidas de catástrofes financieras o empobrecimiento asociados al hecho de tener que pagar por ellos” (FIFARMA, 2019).
Este precepto promulgado por la OMS es adoptado por la legislación colombiana con mayor énfasis sobre la equidad para población vulnerable, sin diferencias injustas en todo el territorio nacional y de acuerdo con las necesidades propias de cada territorio (OPS, 2023).
En este sentido, la sostenibilidad del SGSSS es entendida como la capacidad de contar con los recursos económicos, humanos, ambientales y sociales suficientes que, usados de manera eficiente, garanticen el acceso equitativo y seguro a los servicios y tecnologías en salud de toda la población colombiana, mejorando el bienestar de la sociedad y minimizando los impactos negativos sobre el medio ambiente.
Sistemas de información
Para la toma de decisiones informadas dentro del SGSSS se requiere contar con datos de calidad y confiables, mediante desarrollo de sistemas de información integrados, interoperables, seguros y públicos.
Para Vega-Perez un sistema de información es un conjunto formal de procesos que, operado sobre una colección de datos estructurados de acuerdo con las necesidades de la empresa, recopila, elabora y distribuyen selectivamente.
Así mismo, se ha entendido al sistema de información como el conjunto de personas, datos, información, herramientas de procesamiento y almacenamiento de información dentro de una organización a través de las Tecnologías de la Información y Comunicación (Vega-Perez et al., 2017). Definiciones menos recientes, como la de Laudon & Laudon presentan el sistema de información como un conjunto de componentes interrelacionados que recolectan (o recuperan), procesan, almacenan y distribuyen información para apoyar los procesos de toma de decisiones y de control en una organización (Laudon, 2004).
Adicionalmente, es importante aclarar que en el presente documento “información”, corresponde a aquellos datos que se han procesado significativamente y son útiles y comprensibles para las personas (Laudon, 2004) mientras que los “datos” son la representación en bruto de un evento y son el material de partida para producir información (Laudon, 2004). Bajo este entendido, es preciso afirmar que los datos por sí solos no son útiles, estos deben ser “traducidos” en productos de información y conocimiento para mejorar la toma de decisiones.
Específicamente, en lo referente a los DM, que en conjunto con los medicamentos son los productos más transados en el sector salud, la recolección de datos que se relacionan con el precio, la evaluación de tecnología, el suministro, la calidad, el uso y la disposición final de estas tecnologías en salud contribuyen a generar información y conocimiento suficiente para usar racionalmente los recursos destinados a ellos. De aquí, la relevancia que tiene la disponibilidad de sistemas de información ajustados a la realidad dentro de los sistemas sanitarios.
Lo anterior aportará en el cumplimiento de lo establecido en la Ley Estatutaria 1751 de 2015, sobre la Política de Innovación, Ciencia y Tecnología en Salud “orientada a la investigación y generación de nuevo conocimiento en salud, la adquisición y producción de las tecnologías, equipos y herramientas necesarias para prestar un servicio de salud de alta calidad que permita el mejoramiento de la calidad de vida de la población”.
Ciclo de vida
Según estándares internacionales toda tecnología o producto o dispositivo tiene un ciclo de vida, al respecto la OMS propone una amplia definición del ciclo de vida que abarca fases que van desde la concepción y desarrollo de una idea, la fabricación, empaquetado y etiquetado del producto final, la publicidad para su comercialización, su utilización y su eliminación en el servicio clínico (Cheng, 2003).
Se entiende entonces que el ciclo de vida para los DM, en el marco de este documento, abarca todas las fases que van desde el diseño, la investigación, innovación y desarrollo pasando por el uso clínico, el mantenimiento, cuando aplica, y la disposición final del mismo.
Investigación y desarrollo e Innovación
La investigación y el desarrollo experimental se definen como el trabajo creativo y sistemático realizado con el objetivo de aumentar el volumen de conocimiento incluyendo el conocimiento de la humanidad, la cultura y la sociedad y concebir nuevas aplicaciones a partir del conocimiento disponible (OCDE, 2015).
La innovación se define como el conjunto de actividades científicas, tecnológicas, financieras y comerciales que permiten: introducir nuevos o mejorados productos en el mercado nacional o extranjero, introducir nuevos o mejorados servicios e implantar nuevos o mejorados procesos productivos o procedimientos (Escobar, 2000). La innovación puede tener tres partes: i) la generación, desarrollo y adaptación de una invención, ii) la realización o materialización y iii) la implementación de dicha invención (OCDE, 2018).
La investigación, el desarrollo y la innovación (I+D+i) como se encuentran definidas anteriormente, no solo aumentan el volumen del conocimiento, sino que amplían la gama de opciones diagnósticas y terapéuticas con nuevos o mejorados DM que contribuyen a mejorar la calidad de vida de las personas cuando se logra la implementación de estos en el país, adicionalmente, se convierte en una herramienta de competitividad nacional en materia de esta tecnología en salud.
En desarrollo de los lineamientos internacionales, específicamente sobre la fase de investigación y desarrollo de la OMS (OMS, 2012), y de la política de soberanía en la producción para la seguridad sanitaria (Resolución número 1411 de 2022), el país actualmente cuenta con la Comisión Intersectorial para el Desarrollo y Producción de Tecnologías Estratégicas en Salud (Decreto número 1099 de 2022) con lo cual el país busca estimular la cooperación para la producción local de tecnologías estratégicas.
De otro lado, cabe mencionar que la Política de Ciencia Tecnología e Innovación (CTI) aporta de igual manera a las iniciativas I+D a través de grupos de investigación y universidades, con énfasis sobre el balance de los factores ambientales, sociales y económicos asociados a la producción de tecnologías en salud.
Evaluación de tecnologías en salud (ETES)
La OMS a través de la Resolución WHA CSP28.R9 insta a los países miembros a que (i) “promuevan la creación de procesos decisorios para la incorporación de tecnologías sanitarias basadas en la ETES, lo que puede incluir la seguridad, la eficacia, el costo y otros criterios pertinentes”; y a que (ii) “promuevan el uso de la ETES a fin de fundamentar las políticas de salud pública, incluidas las decisiones sobe la cobertura de los sistemas públicos de salud y la elaboración de guías y protocolos clínicos para las nuevas tecnologías” (OPS, 2012).
La ETES es el proceso sistemático de valorización de las propiedades, los efectos y los impactos de la tecnología sanitaria; debe contemplar las dimensiones médicas, sociales, éticas y económicas y tiene como principal objetivo aportar información para que sea aplicada a la toma de decisiones en el ámbito de la salud. Estas evaluaciones se enfocan en aspectos cómo nivel de beneficios y eficacia, seguridad clínica y técnica, y relación costo-efectividad (OPS y OMS, 2021).
Regulación de precios
Ahora bien, dentro de los mecanismos de intervención del mercado, las políticas de precios farmacéuticos se definen como un “conjunto de principios o requisitos escritos para administrar los precios de productos farmacéuticos, acordados o adoptados por una institución pública (por ejemplo, un gobierno), un grupo de organizaciones compradoras o servicios de salud individuales” (OMS, 2020), concepto aplicable también para DM.
Los objetivos generales de las políticas de precios deben centrarse explícitamente en lograr un acceso asequible y equitativo a productos de calidad para consumidores y sistemas de salud, que deben garantizar una relación calidad-precio basada en mejores resultados de salud a nivel poblacional, además de mantener la seguridad del suministro de productos de alta calidad (OMS, 2020), concepto aplicable también para DM.
La OMS incluye como políticas de intervenciones de precios para las tecnologías en salud: 1) Precio de referencia externo; 2) Precios de referencia internos; 3) Precios basados en el valor; 4) Regulación de márgenes en toda la cadena de suministro y distribución farmacéutica; 5) Promoción de la transparencia de precios; 6) Licitación y negociación; 7) Promoción del uso de medicamentos genéricos y biosimilares de calidad garantizada; 8) Adquisiciones mancomunadas; 9) Fijación de precios de costo-plus para fijar el precio de los productos farmacéuticos; 10) Exenciones o reducciones de impuestos para productos farmacéuticos (OMS, 2020), muchas de estas de aplicación para DM.
Calidad y seguridad
Ya se han mencionado conceptos claves relacionados con la gobernanza, la información, la investigación, el acceso, los precios, pero todo esto debe ir ligado con la calidad, sin embargo, frente a este concepto diferentes autores, entre ellos Voury y De Geyndt, aluden lo complejo de la definición de calidad y reconocen que, al existir ambigüedades sobre este concepto, esta es percibida y definida de formas diferentes (Voury y De Geyndt, 2008).
Vanormelingen por ejemplo, expresa que la calidad es la satisfacción de las necesidades de los usuarios, con soluciones técnicamente óptimas, mientras que Zeithmal define la calidad de un servicio como la discrepancia entre las expectativas y las percepciones de los usuarios (Suplemento, 2008).
La calidad, desde otro punto de vista, puede significar o estar relacionada con la durabilidad de un producto, el precio correcto, servicio oportuno, disponibilidad del servicio, buena aceptación, cumplimiento de estándares altos, satisfacción de las necesidades (Suplemento, 2008).
Este concepto ha evolucionado y se define en la ISO 8402 como la totalidad de características de una entidad que influyen en su capacidad para satisfacer necesidades expresadas e implícitas (ISO, 1995). También se ha ampliado el concepto a la calidad total, siendo esta filosofía la que le permitió a Japón lograr sus niveles actuales de competitividad (Fernandez, 1998).
En el sector salud, algunas definiciones como la de Gilmore y Novaes consideran la calidad como concepto clave para este sector y la definen incluyendo cinco aspectos: (i) un alto nivel de excelencia profesional, (ii) uso eficiente de los recursos, (iii) un mínimo de riesgo para el paciente, (iv) un alto grado de satisfacción por parte del paciente y (v) impacto final en la salud (Gilmore. C., 1996), (Burmester, 1997).
Se entiende entonces por calidad en los DM a las características que satisfacen las necesidades de los usuarios exhibiendo especificaciones que dan cumplimiento a estándares, que prestan mayores beneficios y mitigan los riesgos para la población.
Otro concepto que presenta múltiples interpretaciones y disparidad es “seguridad”, en común se asocia a algo seguro el cual está libre y exento de riesgo u ofrece confianza (RAE, 2021). El término asociado a las tecnologías en salud se encuentra en la literatura definiciones como el “juicio sobre la aceptabilidad del riesgo (medida de la probabilidad de un resultado adverso y su gravedad) asociado al uso de una tecnología en una situación concreta” (IETS, 2014).
Por lo tanto, la seguridad de los DM en este contexto puede definirse como lo hace el Decreto número 4725 de 2005 estableciéndola como la característica de un DM que permite su uso sin mayores posibilidades de causar efectos adversos (Decreto número 4725 de 2005).
Vigilancia Posmercado
El sistema de vigilancia postmercado de los DM actualmente es gestionado operativamente por el INVIMA y a nivel departamental y distrital por las Secretarías Departamentales y Distritales de Salud. El mencionado sistema se aterriza a través de los programas de Tecnovigilancia (Resolución número 4816 de 2008) y Reactivo vigilancia (Resolución número 2013038979 de 2013 modificada en 2020 por la Resolución número 2020007532 expedidas ambas por el INVIMA) que tienen como objetivo identificar, recolectar, evaluar, gestionar y divulgar tanto los riesgos como los efectos y eventos serios o indeseados desconocidos que son producidos por el uso de los DM+RDIV (Minsalud, 2021).
Los agentes del sistema de vigilancia postmercadeo en sus dos programas, se encuentran clasificados por niveles así:
El Nivel Nacional integrado por el MSPS y el INVIMA.
El Nivel Departamental y Distrital, integrado por las diferentes Secretarías Departamentales y Distritales de Salud; en Reactivovigilancia se incluyen además las municipales.
El Nivel Local, integrado por los fabricantes e importadores de DM, RDIV y prestadores de servicios de salud e incluye también los usuarios de DM o cualquier persona que tenga conocimiento de un evento o incidente adverso. En Reactivovigilancia se incluyen además los bancos de sangre, bancos de componentes anatómicos, laboratorios de salud pública, los de referencia, los especializados y de docencia.
Gestión de Tecnologías
La gestión de tecnologías en salud (GTS) es definida por Vilcahuamán y Rivas como “un abordaje sistemático y cuantificable para asegurar que la relación costo/efectividad, eficiencia, seguridad y tecnología disponible sea lo apropiado para cubrir con calidad la demanda por el cuidado de los pacientes” (Rivas, 2006). La GTS se enfoca en los DM y la OMS menciona que aborda aspectos que van desde la evaluación de las necesidades hasta su uso seguro. (OMS, 2012).
Según la OMS, la GTS comprende la planificación, la evaluación de las necesidades, la selección, la adquisición, donaciones, inventario, instalación y mantenimiento de equipos médicos, formación para un uso seguro y disposición final (OMS, 2017). Se menciona también que cada uno de estos ítems abarca una amplia una gama de actividades, que incluyen “proporcionar asesoramiento técnico, planificación y cálculo de costos, seguimiento de contratos, cadena de suministro, disposición final, gestión del personal, gestión de inventario, control de stock de repuestos, consumibles, manejo de residuos e implementación de protocolos de seguridad” (Kaur M et al., 2005).
DIAGNÓSTICO
Contexto nacional de los DM
En Colombia, la regulación de los DM se encuentra a cargo del MSPS a través de la DMTS, que tiene la competencia de “formular, adoptar y evaluar la política farmacéutica, de medicamentos, de dispositivos, de insumos y tecnología biomédica, y establecer y desarrollar mecanismos y estrategias dirigidas a optimizar la utilización de los mismos” (Decreto número 4107 de 2011). Esta regulación del sector de Dispositivos Médicos en los últimos años ha mostrado avances importantes frente a la política de precios y en la definición del estándar semántico, entre otros.
De acuerdo con la información de la base general de establecimientos que reposa en el INVIMA con corte a 31 de diciembre de 2022, en cuanto a establecimientos de DM, así como de RDIV se evidencia que el 80% corresponden a fabricantes internacionales y el 20% a fabricantes nacionales.
El crecimiento de establecimientos en el periodo 2014 a 2022 para los fabricantes de DM nacionales fue del 220% y para los importadores del 172% con un promedio de crecimiento general de 523 establecimientos. Los establecimientos de RDIV por su lado, tuvieron un incremento del 127% para los importadores y un 140% para los nacionales en el mismo periodo de tiempo, con un promedio de crecimiento general de 28 establecimientos.
Ahora bien, la balanza comercial del sector de los DM en Colombia entre enero de 2007 a diciembre de 2021 es negativa, puesto que, el valor de las importaciones supera el de las exportaciones. En cuanto a las cifras reportadas en el año 2021 de productos importados fue por valor de 1.629.897 millones de dólares, mientras que para los productos exportados representaron únicamente 107.515 millones de dólares.
Gráfico 1. Balanza Comercial (Ene 2007 – Dic 2021) sector de los Dispositivos Médicos en Colombia

Fuente. CVN - Centro Virtual de Negocios. Tomado de Libro Comercio. Cámara dispositivos Médicos - ANDI.
La comercialización de DM y RDIV en Colombia, se realiza bajo registros sanitarios (RS) o permisos de comercialización (PC) otorgados por el INVIMA los cuales, según el riesgo, pueden ser automáticos o con control previo y pueden amparar varios productos siempre y cuando, estos tengan características similares.
Según la base de datos del INVIMA, al 31 de diciembre de 2021, había unos 16.172 registros sanitarios (RS) asociados a DM y equipos biomédicos (EB). De estos, 460 RS estaban vencidos, 86 RS suspendidos, 1.031 RS cancelados y 15.172 RS vigentes. El 91,09% de los RS vigentes correspondía a DM importados y el 8,91% a DM de fabricación nacional. Esto indica que el mercado de DM en Colombia es predominantemente importado.
Gráfico 2. Comparación porcentual del estado de los registros sanitarios de DM y RDIV
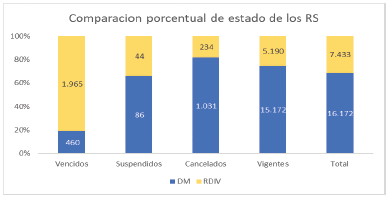
Fuente: elaboración propia a partir de los datos de Invima.
De igual forma, había unos 7.433 RS asociados a reactivos de diagnóstico in vitro (RDIV). De estos, 1.965 RS estaban vencidos, 44 RS suspendidos, 234 RS cancelados y 5190 RS vigentes. El 96,33% de los RS vigentes correspondía a RDIV importados y el 3,67% a RDIV de fabricación nacional. Esto muestra que el mercado de RDIV en Colombia también es mayoritariamente importado.
Gráfico 3. Comparación de RS nacionales e importados de DM y RDIV
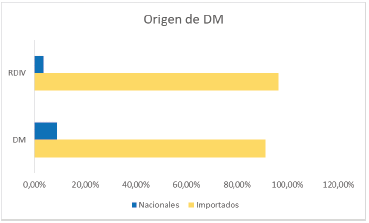
Fuente: elaboración propia a partir de los datos de Invima.
En cuanto al riesgo de los DM y EB con RS vigente, se observa que para los DM el 24,66% de los RS tienen clasificación I y el 43,38% de los RS tienen clasificación IIA. Esto indica que en su mayoría hay RS de riesgo IIA de DM y EB.
En cuanto a la categoría de los RDIV con RS vigente, se observa que para los RDIV el 5,47% de los RS tienen categoría I, y el 48,36%. de los RS tienen categoría II 2510. Esto indica que en su mayoría hay RS de categoría II de RDIV.
Gráfico 4. Clasificación de riesgo de los RS de DM y RDIV
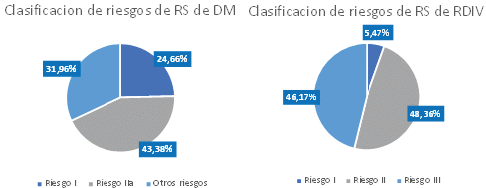
Fuente: elaboración propia a partir de los datos de Invima.
Al realizar una revisión solamente a la fabricación nacional de DM, se tiene que, de los 1.352 RS vigentes, el 7,79% son de riesgo I y IIA, respectivamente.
Referente a la fabricación nacional de RDIV, se tiene que, de los 115 RS vigentes, el 82,6% son de categoría I y II.
Debido a la falta de estandarización de la base, es difícil identificar las clases de DM que se fabrican en el país, sin embargo, al realizar una clasificación uno a uno de los 1352 RS de fabricación nacional asociando por tipologías, se puede identificar que el 98,6% son DM de tipología consumible y el 1,04% corresponde a la tipología de los equipos biomédicos de tecnología controlada.
En cuanto a la frecuencia de expedición de los RS, se evidencia una línea de tendencia al aumento desde el año 2.005; en los últimos 5 años el promedio anual es de 169,25 RS nuevos expedidos de DM y 8 RS nuevos expedidos de RDIV de fabricación nacional.
De la información correspondiente a los RS expedidos por el INVIMA anteriormente expuesta, se concluye que en Colombia se fabrican DM de bajo y moderado riesgo (I y IIA) y de RDIV de bajo y mediano riesgo (I y II), en su mayoría de tipología consumibles y que en promedio se expiden 169,25 nuevos RS de DM y 8 RS nuevos de RDIV anuales de fabricación nacional.
Ahora bien, otro factor para tener en cuenta corresponde al ciclo de vida de los DM que comprende las fases desde la concepción y desarrollo de una idea, hasta la fabricación, empaquetado y etiquetado del producto final, la publicidad para su comercialización, su utilización y eliminación del servicio clínico.
Como se menciona en el documento de Árbol de problemas en el marco del Análisis de Impacto Normativo (AIN) de Investigación Clínica con Dispositivos Médicos (DM) y Reactivos de Diagnóstico In Vitro (RDIV), desarrollado por el MSPS en conjunto con Invima, la investigación clínica hace parte importante del proceso de desarrollo de nuevos dispositivos médicos puesto que permite la obtención de alternativas terapéuticas para el tratamiento de patologías de alto impacto en salud pública. “El objetivo principal de los estudios clínicos es demostrar la seguridad y eficacia de estas potenciales tecnologías sanitarias, siendo el primer eslabón en la cadena productiva de los Dispositivos Médicos” (Minsalud, 2023b).
La adopción de las Buenas Prácticas Clínicas (BPC) dentro de la normativa nacional en el año 2005, visibilizó la investigación clínica colombiana a nivel internacional. También promovió el crecimiento en el número de centros de investigación certificados en el país, llegando en la actualidad a existir 141 centros de investigación clínica y 77 comités de ética certificados por el Invima en medicamentos con corte a agosto de 2022 (Minsalud, 2023b).
En Colombia, según el Decreto número 4725 de 2005, la fabricación, empaquetado y etiquetado del producto final y su publicidad están a cargo del fabricante o del importador, bajo la inspección, vigilancia y control (IVC) del Invima. El uso y la disposición final es realizada por los prestadores de servicios de salud, definidos en la Resolución número 3100 de 2019.
Por otro lado, con relación al talento humano se observa que, en el año 2007, la OMS en la Resolución WHA 60.29 insta a los países miembros a que formulen estrategias de “evaluación, planificación, adquisición y gestión de las tecnologías sanitarias, en particular de los dispositivos médicos, en colaboración con personal dedicado a la evaluación de las tecnologías sanitarias y la ingeniería biomédica”. En este sentido, dado que el desarrollo del ciclo de vida de los DM depende de las acciones acertadas y oportunas de talento humano, se requiere contar con profesionales de las diferentes disciplinas del área de la salud en las fases de evaluación, planificación, regulación, adquisición y gestión de las tecnologías sanitarias, para el uso adecuado y disposición final.
Teniendo en cuenta la diversidad de tecnologías existentes en el mercado, también debe integrarse profesionales de otras disciplinas tales como de las ciencias básicas – física e ingenierías, específicamente provenientes de programa en ingeniería biomédica, bioingenierías y físicos médicos, con el fin de impulsar la innovación y desarrollo de nuevas tecnologías en el país, además, para el apoyo en la gestión durante del ciclo de vida. Estas disciplinas pese a la necesidad en el SGSSS y el mercado de los DM no han tenido una inclusión en sus procesos.
De la búsqueda realizada, se identifica que existen 23 programas de ingeniería biomédica en 18 instituciones de educación superior en diferentes partes del país, los graduados en esta profesión se registran ante el Consejo Profesional Nacional de Ingeniería (COPNIA), en cumplimiento a la Ley 842 de 2003 (Ley 842 de 2003). Al consultar esta entidad se evidencia que desde el año 2000 se han registrado más de 3.800 profesionales para ejercer la ingeniería biomédica, con un crecimiento exponencial a lo largo de los años.
Problema identificado
El problema de la situación de los DM se concreta en:
“Dificultades en el acceso equitativo de la población colombiana a los DM y desintegración de los factores que inciden en la disponibilidad, calidad, continuidad, uso seguro y adecuado en la salud individual y colectiva para contribuir con los resultados en salud que se dan en el Sistema General de Seguridad Social en Salud”.
Causas
1. Carencia de un sistema de información de DM que integre las herramientas existentes e identifique las que se requieran para responder a las necesidades de toma de decisiones en el sector.
La política de Gobierno Digital (Decreto número 767 de 2022) anteriormente denominada Gobierno en Línea, tuvo como logro el salto en la inclusión social y en la competitividad del país a través de la apropiación y el uso adecuado de las Tecnologías de la Información y las Comunicaciones (TIC).
Esta estrategia fue adoptada por el MSPS dando como resultado el Sistema Integral de Información de la Protección Social en Colombia (Sispro), que permite a todos los actores del SGSSS, al Gobierno nacional y a la ciudadanía en general acceder a la información epidemiológica y estadística, y a herramientas de análisis, para facilitar y focalizar la gestión y la toma de decisiones en salud.
En este sentido, y en el marco del Sispro, el MSPS viene desarrollando sistemas de información para cada tecnología, que busca satisfacer las necesidades específicas del SGSSS y facilitar el monitoreo, seguimiento, evaluación y el mejoramiento de la vigilancia y el control, optimizando la toma de decisiones y el logro de resultados en salud.
Por otra parte, el Instituto de Evaluación Tecnológica en Salud (IETS), mediante diagnóstico realizado en 2019, encontró que el sector de DM no cuenta con un sistema de información oficial público que reúna las características de información “periódica, confiable, oportuna, eficiente, disponible, comparable, histórica y completa” que permita la toma de decisiones informadas al conjunto de actores y agentes del SGSSS (IETS, 2019)
Inicialmente, el denominado estándar semántico y codificación de DM reglamentado por la Resolución número 1405 de 2022 y reiterado en las bases del Plan Nacional de Desarrollo 2023-2026, tiene como finalidad la identificación, denominación y clasificación de estas tecnologías facilitando su uso, trazabilidad y el intercambio de información entre actores y agentes involucrados en la comercialización, distribución, inspección, vigilancia y control durante el ciclo de vida y en los diferentes eslabones de la cadena.
También el estándar semántico y codificación de DM permitirá el uso de datos confiables para estudios epidemiológicos, promoción de DM costo-efectivos y prioritarios, investigación, innovación y desarrollo.
Los objetivos son: (i) garantizar la trazabilidad de los DM a lo largo de toda la cadena de comercialización, incluida la investigación, promoción, uso y disposición final, (ii) facilitar el manejo, suministro, adquisición y uso en los procesos de prestación de servicios de salud, actividades de promoción y prevención y el inventario y mantenimiento de los equipos biomédicos, (iii) mejorar la gestión relacionada con el gasto del SGSSS, (iv) facilitar los procesos de regulación y vigilancia de precios y consumo, (v) facilitar el intercambio de información entre las autoridades competentes en materia de inspección, vigilancia y control sanitario y (vi) facilitar la identificación y clasificación según estándares internacionales.
En este punto, se requiere avanzar en la implementación de la Resolución número 1405 de 2022, que estableció un horizonte de tiempo de cuatro años, para que el SGSSS cuente con el estándar semántico operando y establezca mecanismos para la ciberseguridad de la información.
1.1. Difusos requerimientos para el diseño del sistema de información de DM.
Según el IETS, en su diagnóstico de 2019, las perspectivas de las necesidades y requerimientos del sistema de información para DM son diversas según los actores.
Desde la perspectiva de las sociedades participantes, mencionan que “el sistema de información debe garantizar la disponibilidad de información, que incluya información sobre calidad, estrategias de uso y costo-beneficio” (IETS, 2019). Por parte de las entidades territoriales, se menciona que el sistema de información podría estar incluido en SISPRO para facilitar el acceso en diferentes niveles, que apoye las funciones de inspección, vigilancia y control de estos actores, conociendo tanto “la base instalada de tecnologías de alto riesgo por número de habitantes en el país” (IETS, 2019) como las características técnicas, de calidad de las mismas, de manera que el sistema permita realizar “consulta de registros sanitarios y permiso de comercialización tan solo con la información de lote y/o serie de un dispositivo, páginas web para consulta de proveedores e importadores autorizados” (IETS, 2019).
Los prestadores de servicios de salud, desde su perspectiva consideran que el sistema de información debe ser de “fácil uso, acceso abierto, codificado bajo estándares internacionales y que a su vez permitan hacer analítica” (IETS, 2019), ya que para estos actores es significativo realizar sinergia con otras instituciones en salud. Precisan también que sea importante “que contenga datos actualizados y estandarice la información nacional de la documentación de los dispositivos médicos disponibles” (IETS, 2019).
Por su parte las agremiaciones industriales, perciben que “el sistema de información debe considerar las características de cada subsegmento del mercado de los DM para mejorar su entorno” (IETS, 2019).
De esta forma es importante caracterizar las fuentes existentes y consolidar los requerimientos del sistema de información para DM.
1.2. Débil análisis de la información recolectada actualmente sobre dispositivos médicos para aportar en la toma de decisiones de los actores.
Según el Instituto de Evaluación Tecnológica en Salud (IETS), en el diagnóstico realizado en el año 2019, se puede identificar la poca comunicación en temas de DM, hay “falta de información y difusión hacia los actores involucrados” (IETS, 2019), incluyendo en esta parte, la importancia de “comunicar resultados de las evaluaciones de desempeño, así como proporcionar esta información a toda la cadena de actores involucrados” (IETS, 2019).
De otro lado, se cuenta actualmente con información almacenada y gestionada en diferentes fuentes tales como:
- En el Sispro del MSPS se encuentra alojado el Sisdis, el cual permite reportar precios por parte de fabricantes (incluye importadores), intermediarios y prestadores de servicios de salud.
- El Invima cuenta con bases de datos independientes para registros sanitarios, condiciones técnico-sanitarias de fabricantes nacionales, certificado de capacidad de almacenamiento y acondicionamiento de importadores y las de los programas posmercado de tecnovigilancia y reactivovigilancia. Estas bases también están en www.datos.gov.co (datos abiertos).
Externamente al sector de los DM se encuentran fuentes de información que pueden aportar datos de DM tales como la DIAN, la Supersalud, las entidades territoriales de salud y prestadores de servicios de salud.
Sin embargo, son pocos los análisis que se realizan sobre la información que se recolecta y escasa la difusión que se realiza de estos, la cual puede aportar a la toma de decisiones de los actores en los diferentes puntos del ciclo de vida de los DM.
2. Insuficientes lineamientos que contribuyan al fortalecimiento de la investigación, el desarrollo y la práctica clínica en materia de DM.
2.1. Escasos espacios de discusión sobre la investigación y desarrollo (I+D) con los actores involucrados de DM.
Dentro del diagnóstico realizado por el IETS, uno de los principales problemas identificados es “la ausencia de redes de gestión para compartir el conocimiento, donde los proyectos e innovaciones se visibilicen y puedan generar un interés en un promotor patrocinador” (IETS, 2019) ya que, si bien existen desde la academia algunas redes, es escaso aún el enlace con patrocinadores y alianzas público-privadas que fortalezcan la I+D+i con relaciones basadas en la confianza del triángulo Estado-academia-industria.
Dentro de los actores que identifica el IETS en su documento para la articulación y ampliación del mencionado triangulo están la academia, los prestadores de servicios de salud, los entes reguladores, el sector industrial y el gobierno.
Por lo que se requiere la creación de espacios de discusión sobre la investigación y desarrollo (I+D) con los actores involucrados de DM.
2.2. Deficientes lineamientos sobre investigación y desarrollo (I+D) así como de práctica clínica en DM.
La investigación en Colombia si bien se encuentra regulada y cuenta con organismos que la impulsan, ha tenido mayores avances en sectores diferentes a la salud, no obstante, el sector farmacéutico con presencia de industria nacional viene produciendo medicamentos de síntesis química y antiveneno, y se proyecta a futuro en la producción de radiofármacos, en consonancia impulsa y desarrolla investigación clínica (Molina, Botero y Giraldo, 2016).
Algunas universidades y grupos de investigación, en coordinación con Minciencias, adelantan proyectos de investigación de DM de carácter incremental, como es el caso de lo sucedido en el marco de la pandemia por el COVID-19, donde se presentaron iniciativas de fabricación, por ejemplo de ventiladores, en donde algunas universidades trabajando en conjunto con hospitales presentaron prototipos nacionales, los cuales por la falta de lineamientos claros sobre los requisitos que deben cumplir en el momento de someter los protocolos de investigación al Invima para el desarrollo de investigación clínica con DM, no llegaron a finalizar el proceso. Los estudios clínicos sobre DM pueden ser utilizados por quienes hagan investigación o desarrollo de DM en innovación incremental o de novo.
En el sector de los DM colombianos, además, se tienen identificados algunos problemas como: “ausencia de recursos financieros para hacer investigación y desarrollo local de tecnologías y dispositivos médicos” (IETS, 2019), la falta de gestión de los recursos existentes (IETS, 2019) y un factor importante que la investigación no responde a necesidades del perfil epidemiológico (IETS, 2019), lo que retrasa aún más el desarrollo y producción de tecnologías nacionales, razón que conduce a la idea de buscar la adecuación de un ecosistema que permita el desarrollo de investigaciones clínicas con DM de alta calidad en coordinación con actores y agentes del SGSSS.
Colombia, en cuanto a tecnologías sanitarias, contaba a 2016 con un registro de 927 estudios clínicos, aunque el dominio del mercado mundial de estudios clínicos seguirá liderado por los países de altos ingresos (Molina, Botero y Giraldo, 2016).
En los últimos años, se ha encontrado un aumento en el interés de desarrollar ensayos clínicos en el país (Minsalud, 2023b). Este interés se ha visto incrementado de manera proporcional con el número de sometimientos de protocolos con esta tecnología sanitaria ante el INVIMA, por lo que se cuenta con información incipiente, por esta razón se hace vital la adecuación de un ecosistema que permita el desarrollo de investigaciones clínicas con DM de alta calidad (Minsalud, 2023b).
Como se muestra en el gráfico siguiente, la solicitud de estudios clónicos que se allegan a la sala especializada de DM y RDIV del Invima ha incrementado en un 56,5% desde el año 2022 hasta el mes de septiembre de 2023.
Gráfico 5. Total, trámites vs. estudios clínicos, conceptuados por la sala especializada de DM y RDIV en el periodo 2018-2023 (septiembre)

Fuente: datos suministrados por Invima.
En el caso de Colombia, la regulación vigente para el desarrollo de investigación en salud con seres humanos está definida por la Resolución número 8430 de 1993”, por la cual se establecen las normas científicas, técnicas y administrativas para la investigación en salud, que dicta los lineamientos generales para realizar investigación con seres humanos incluyendo nuevos recursos profilácticos, de diagnóstico, terapéuticos y de rehabilitación, señalando algunos requisitos éticos, técnicos y legales transversales (Minsalud, 2023b).
Así mismo, la Resolución número 2378 de 2008 define los requisitos para la certificación en Buenas Prácticas Clínicas a las instituciones en salud que estén interesadas en desarrollar investigación clínica con medicamentos (Minsalud, 2023b).
No obstante, a pesar de que estas normas han permitido definir un marco general para el desarrollo de investigaciones con seres humanos, han ido quedando desactualizadas, debido principalmente a los avances de la investigación, el desarrollo de nuevos diseños de ensayos clínicos y la introducción de las tecnologías de la información (Minsalud, 2023b).
Se evidencia la necesidad de contar con una agenda regulatoria sobre investigación y desarrollo (I+D) y práctica clínica en DM, basados en normas nacionales y armonizadas con las normas internacionales existentes, para asegurar la solidez científica y ética de la investigación.
3. Debilidades en la rectoría y gobernanza sobre los DM con escasas capacidades técnicas en los actores.
Dentro del diagnóstico realizado por el IETS, los encuestados mencionaron que “deben estar articulados todos los actores intersectorialmente y que se tenga mayor presencia del Ministerio de Salud y Protección Social” (IETS, 2019).
Según el IETS, con el panorama actual de la gobernanza en el nuevo orden global, se hace evidente que quien regula no posee el monopolio del conocimiento, de la experiencia y de los recursos necesarios para resolver los problemas, por lo tanto, debe ingeniar y reinventar nuevas formas para generar entre todos las políticas públicas, en compañía de otros actores estratégicos, como la ciudadanía (IETS, 2019).
De otro lado, se observa baja capacidad en las entidades territoriales relacionada con su estructura tanto técnica como de talento humano, que limita el desarrollar una efectiva IVC. Téngase en cuenta, por ejemplo, el artículo 65 del Decreto número 4725 de 2005, en donde establece que estas entidades en coordinación con Invima ejercerán las acciones de inspección, vigilancia y control sanitario y aplicará las medidas de prevención y correctivas necesarias para dar cumplimiento a las disposiciones. En este sentido se requiere estrategias que fortalezcan el rol de las entidades territoriales de salud para la fiscalización sanitaria de los DM.
Finalmente, el IETS en el resumen ejecutivo aborda un aspecto que va en línea directa con la gobernanza, como es la rectoría en salud, con la afirmación “La regulación de los DM se encuentra a cargo del Ministerio de Salud y Protección Social a través de la Dirección de medicamentos y tecnologías en salud, quienes tienen responsabilidad de 'formular, adoptar y evaluar la política farmacéutica, de medicamentos, de dispositivos, de insumos y tecnología biomédica, y establecer y desarrollar mecanismos y estrategias dirigidas a optimizar la utilización de los mismos”.
Teniendo en cuenta que la gobernanza en el SGSSS es transversal y permea las estrategias contenidas en las políticas de salud, su abordaje determina la concreción de los elementos de legitimidad, estabilidad y sostenibilidad, los cuales son necesarios para que los objetivos de estas políticas sean realizables, así como, definir los roles de los agentes responsables de su implementación y ejecución, y establecer los mecanismos para promover el mejoramiento continuo a través de una eficaz gestión de los DM.
La rectoría y gobernanza regulatoria tendrán en cuenta las obligaciones y compromisos internacionales suscritos y que tienen fuerza vinculante para Colombia.
4. Escasas estrategias para fortalecer la disponibilidad y la asequibilidad de DM priorizados, que respondan al perfil epidemiológico de la población colombiana.
4.1. Escaso desarrollo del componente de disponibilidad de los dispositivos médicos.
El IETS en su documento menciona que el abastecimiento puede verse impactado algunas veces por “los procesos engorrosos de importación” (IETS, 2019). Así mismo, al relacionar disponibilidad con el uso, se menciona que “la utilización de los DM está ligada a la disponibilidad en el mercado, que es reducida o controlada para todos los niveles de atención en salud y en los diferentes territorios”.
En cuanto a las dificultades de abastecimiento de DM se encuentra que el país vivió este fenómeno en tiempo de pandemia y algunos rezagos posteriores debido a la baja disponibilidad de DM y la escasa fabricación nacional. Esto fue posiblemente causado porque los países fabricantes reservaron su producción para consumo propio (Union Medical, s.f.), al igual, que el apoyo y fomento de empresas locales era deficiente y no existía suficiencia de materias primas a nivel mundial. Según lo informado por la ANDI, este fenómeno responde a variables internacionales como la relocalización de las plantas de producción, lo cual no es un tema exclusivo para nuestro país.
Si bien el país ha presentado riesgo de escasez no ha llegado al desabastecimiento de DM que significaría fallas en el acceso para la atención, sumado a lo anterior cabe reconocer que hacen falta herramientas de identificación de estas tecnologías prioritarias que den respuesta al perfil epidemiológico del país, así como el fomento de producción nacional para el autoabastecimiento.
Cabe destacar finalmente que el SGSSS requiere que todas las tecnologías sanitarias, en especial los DM, cuenten con evaluación para el uso seguro, que sea de calidad, requerido por el SGSSS, disponible en el mercado local y a precios asequibles.
De esta forma se requiere una herramienta de monitoreo del estado del abastecimiento de DM que permita aumentar y gestionar la disponibilidad de estos, además de planteamiento de estrategias con los actores para evitar el riesgo de escasez de DM en Colombia.
4.2. Incipientes avances en la evaluación de tecnologías (ETES) en dispositivos médicos.
Colombia cuenta con el IETS, que desde 2012 ha desarrollado más de 1.000 productos relacionados con la revisión de la evidencia, la economía, la analítica, la actuaria, los métodos cualitativos y la investigación social en salud, entre los cuales se encuentran evaluaciones de DM, muchos de estos hoy contemplados en el Plan de Beneficios, contribuyendo así al acceso a la población de tecnologías seguras y con evidencia científica.
En la encuesta realizada por el IETS, los participantes señalaron que no existe suficiente divulgación sobre los resultados de la Evaluación de Tecnologías Sanitarias -ETES de DM, ni apropiación de las metodologías existentes (IETS, 2019). En este punto, enfatizaron en que no se conocen los lineamientos o pautas para estas ETES, no existen guías para hacerlas a nivel micro ni procesos de capacitación para una adecuada interpretación de resultados de las ETES de DM (IETS, 2019). Es pertinente indicar que el IETS tiene un manual metodológico para la elaboración de evaluaciones de efectividad, seguridad y validez diagnóstica de tecnologías en salud (IETS, 2014), que probablemente no ha tenido una buena difusión para el sector de DM.
En la encuesta realizada también se encontró que se asocia el concepto de ETES con vigilancia y tecnovigilancia, evidenciando una confusión en la población general en la terminología, además explica la “necesidad de articular los procesos, es decir, garantizar que la información obtenida desde la tecnovigilancia sirva para el desarrollo de evaluaciones sobre DM” (IETS, 2019).
De igual forma, los participantes en la encuesta expresan que los “procesos de evaluación deben servir para tomar decisiones informadas y adoptar recomendaciones basadas en evidencia” (IETS, 2019). En este punto, se señaló como un problema reiterativo “la inexistencia o debilidad de los comités institucionales para evaluación de tecnologías; los que existen, además, solo se enfocan en criterios de costos, dejando de lado la necesidad de garantizar la integralidad y la calidad en la atención en salud” (IETS, 2019).
Por otra parte, los participantes mencionaron que “la evaluación se concentra en los aspectos farmacéuticos primordialmente, y no consideran la necesidad de tener evaluaciones de nuevas tecnologías, o evaluaciones de nuevas aplicaciones de las tecnologías existentes” (IETS, 2019), consideran entonces que, en el marco de las ETES se deja a un lado los DM.
Es importante resaltar de la encuesta realizada por IETS que el 29% de las aseguradoras y el 23% de los prestadores encuestados no cuentan con comité de evaluación de DM, y una pequeña proporción, el 14% de Aseguradores y el 6% de prestadores, manifestaron no reunirse, como se puede ver en el gráfico 6. Dado esto es importante implementar estrategias que impulsen e incentiven a las organizaciones a la realización de comités de ETES teniendo en cuenta su importancia para la toma de decisiones en salud (IETS, 2019).
De manera similar a lo manifestado durante la encuesta, en el espacio participativo realizado por el IETS en el marco de la realización del diagnóstico hecho en 2019, se llamó la atención a que tampoco “existe análisis multicriterio que incorpore usos inadecuados, afectación de los pacientes y no solo recursos” (IETS, 2019) y que, por ejemplo, el escaneo de horizonte se realiza solo en casos aislados para invertir. Así mismo en las ETES realizadas “tampoco se incorporan los aspectos de seguridad del paciente” (IETS, 2019), el proceso se realiza de manera empírica y es incipiente, y también se denota poco interés por aprender (IETS, 2019).
Gráfico 6. Periodicidad de reunión del comité de ETES
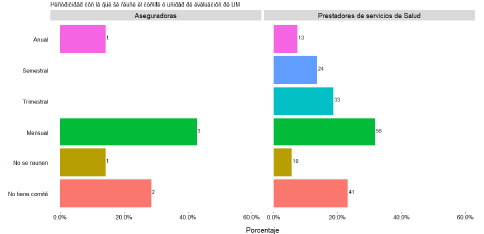
Fuente: diagnóstico del sector de DM (IETS, 2019).
4.3. Escaso desarrollo de la política de precios para DM.
Respecto a la asequibilidad, el IETS menciona que se debe buscar transformar el cuidado a partir de la medicina basada en la evidencia y la economía de la salud, buscando apoyar las compras de DM por resultados, más no por precio y que en el país no hay criterios de costo efectividad sino de ahorro de costos (IETS, 2019). También destacan que hay altos costos de la tecnología y algunos actores consideran importante regular el costo, tal como están regulados los medicamentos (IETS, 2019). Así mismo refieren que aún hay escaso fomento para que el país cuente con tecnología a buenos precios que facilite la atención a los pacientes con alta tecnología (IETS, 2019).
En cuanto al reporte de precios, en el gráfico 7 se observa la percepción de los participantes en la encuesta con respuestas muy heterogéneas, de donde llama la atención que el 46% de los encuestados consideran necesario incluir todos los DM en el control de precios; el 29% consideran que se pueden priorizar, el 21% no conocen sobre este tema y el 4% mencionan que no se debe reportar precios de DM.
Teniendo en cuenta que los actores mencionaron un elevado costo de algunos de los DM (sobre todo, en las regiones más apartadas del país), se identifica la necesidad de incluir en el componente de regulación de precios de los DM variables como la accesibilidad por cuestiones geográficas en el contexto del mercado colombiano de DM a efecto de que sea una herramienta para incentivar la libre competencia y disminuya las asimetrías de información al momento de realizar los procesos de adquisición (IETS, 2019).
El diagnóstico del IETS refiere que la regulación de precios debe ser un proceso consensuado, pues se tiende a dejar de producir y comercializar cuando es impuesto; en otras palabras, debe existir la regulación de precios, pero de manera estructurada conociendo lo que se tiene y con un adecuado estándar semántico e indican que los precios deben estar establecidos por referenciación de precios internacionales en países con sistemas maduros en salud (IETS, 2019).
Gráfico 7. Percepción sobre reporte de precios
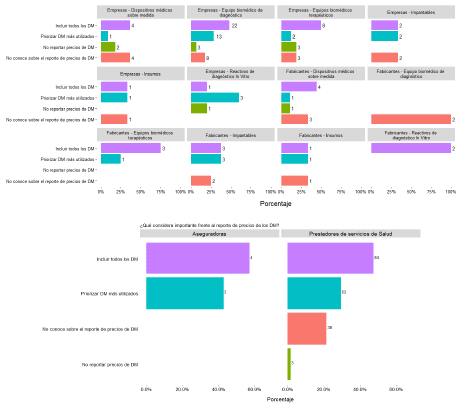
Fuente: Diagnostico del sector de DM (IETS, 2019).
Finalmente, como resultado de la política de precios para DM, actualmente se tienen seis mercados relevantes en régimen de libertad vigilada (únicamente reporta el precio de comercialización): DM endovasculares coronarios, anticonceptivos, implantables auditivos de conducción ósea, implante para córnea, implante coclear, DM implantables para terapia cardiaca de alto voltaje (desfibriladores y cardio-resincronizadores) y tan solo un DM, estent coronario medicado, en control directo de precios (establece un precio máximo de venta).
4.4. Escasa producción local de DM.
En el marco de la implementación de la política de soberanía en la producción para la seguridad sanitaria, el país conformó la Comisión Intersectorial para el Desarrollo y Producción de Tecnologías Estratégicas en Salud (Decreto número 1099 de 2022) con la que se busca estimular la cooperación entre actores para la producción local de tecnologías estratégicas.
Como ejemplo de esto se encuentra la participación público-privada en la estrategia para la producción local de vacunas y la cooperación con organismos internacionales para establecer la viabilidad de producción local de radiofármacos con el Organismo Internacional de Energía Atómica (OIEA).
El panorama de DM muestra que este sector en el país, fabrica DM de algunas tipologías de los riesgos alto y medio, y con mayor proporción de bajo riesgo, con lo cual no auto abastece las necesidades de atención en salud, llevando a que aproximadamente el 97% de los DM sean importados. Más aún, en concordancia a este panorama la investigación clínica no ha tenido desarrollo.
Otro factor que determina la baja producción nacional según la industria de RDIV, son los altos costos de las materias primas y de los “Gold estándar”, es un estándar o patrón de oro para validación de sus pruebas, asociado a los escasos laboratorios certificados en el país (Minsalud, 2021).
Como se indicó en el contexto nacional, solo el 38% del volumen total de fabricantes de DM presentes en el mercado colombiano, son nacionales y se centra en los DM de menor riesgo sin desconocer que hay fabricación de DM en los otros niveles de riesgo (IIa, IIb y III) (Minsalud, 2019) como se muestra en la tabla 2. En RDIV el 43% son nacionales y producen generalmente riesgo I y II (Minsalud, 2021).
Tabla 1. DM de producción nacional
| Riesgo I | Riesgo IIA | Riesgo IIB | Riesgo III |
| Venda de algodón o elástica | Set de infusión de líquidos | Sistemas de implantes osteosíntesis | Suturas absorbibles y sintéticas |
| Esparadrapo | Desinfectantes | Suturas de diferentes materiales no absorbible | Matriz extracelular |
| Campos médicos | Sondas de alimentación incluyendo la radiopaca | Implante dental | Membrana de colágeno para regeneración tisular |
Fuente: Elaboración propia Grupo Dispositivos Médicos 2018.
Por otra parte, desde el año 2023, el Ministerio de Comercio, Industria y Turismo se encuentra trabajando en la Política de Reindustrialización para transitar de una economía extractivista a una economía del conocimiento, productiva y sostenible (Mincit, 2023. Los objetivos de esta política se concentrarán en cerrar brechas de productividad, fortalecer los encadenamientos productivos y la inversión, diversificar y sofisticar la oferta interna y exportable, y profundizar la integración con América Latina y el Caribe (Mincit, 2023).
Dentro de esta política se encuentra la apuesta estratégica sobre la reindustrialización en el sector salud, donde se menciona que Colombia “generará capacidad de producción local de excipientes activos, medicamentos, vacunas, dispositivos y partes para dispositivos médicos” (Mincit, 2023), por lo que las acciones intersectoriales que se presenten en este marco serán definitivas para el sector y para la producción local de DM basados en altos estándares teniendo en cuenta las obligaciones y compromisos internacionales suscritos y que tienen fuerza vinculante para el país.
5. Débiles lineamientos postmercado, regulación y reglamentación técnica que hagan énfasis en la promoción de la calidad y seguridad de los DM.
La calidad y seguridad como se menciona en el marco teórico, son características de los DM que impactan de manera positiva la atención de los pacientes, por lo tanto, a continuación, se mencionarán los temas del sistema de vigilancia postmercado, así como de regulación y reglamentación técnica de los DM.
5.1. Debilidades en la capacidad técnica de los agentes del sistema de vigilancia postmercado de los Dispositivos Médicos.
A pesar de los esfuerzos del Invima en el fortalecimiento de los programas de Tecnovigilancia y Reactivovigilancia, aún se evidencian debilidades en la capacidad técnica de los actores del sistema de vigilancia postmercado de los DM, tales como la continua rotación de los referentes territoriales de salud que no permite el trabajo continuo y el desarrollo de experticia en el manejo de los reportes lo cual conlleva a la improvisación, bajo registros y la deficiente retroalimentación.
Según el IETS, se debe facilitar la retroalimentación a los interesados en los programas de vigilancia postmercado, así como, evitar el temor a los reportes de eventos adversos, informando a los usuarios que los programas cuentan con política de confidencialidad (IETS, 2019).
Frente a los eventos e incidentes adversos asociados a los DM, el Programa Nacional de Tecnovigilancia del Invima, informa que en el periodo comprendido entre 2015 y 2019, se notificaron 53.391 reportes, de los cuales 18.168 corresponden a problemas de fabricación que provocaron 4.609 eventos adversos y 13.559 incidentes.
Por su parte, al Programa de Reactivovigilancia se notificaron en el mismo periodo 1.552 reportes, de los cuales 159 están asociados a la fabricación, que produjeron 141 incidentes adversos, 6 eventos adversos y 12 sin clasificar.
La evolución de Programa Nacional de Tecnovigilancia no se ha limitado a vigilar los eventos reportados mediante la estrategia de reporte espontáneo (Vigilancia Pasiva), exige fomentar el desarrollo de un Sistema Integral de Tecnovigilancia, que considere la vigilancia proactiva (Enfoque de Riesgo) y establecer estrategias para desarrollar la vigilancia activa/intensiva e investigación específica de los dispositivos médicos. (García-Vega, 2015).
De igual forma a nivel internacional, el IMDRF cuenta con el documento “IMDRF terminologies for categorized Adverse Event Reporting (AER): terms, terminology structure and codes” desde el año 2020.
Adicionalmente, el MSPS con el fin de fortalecer la Inspección, Vigilancia y Control (IVC), de los productos de uso y consumo humano expidió la Resolución número 1229 de 2013, por la cual se establece el modelo de inspección, vigilancia y control sanitario para los productos de uso y consumo humano, que conmina a que para las tecnologías en salud se diseñe un modelo de IVC sanitario que permita operativizar las acciones en el territorio nacional, con el concurso de las entidades de orden nacional y territorial encargadas.
5.2. Debilidad en la regulación y la reglamentación técnica de los DM entorno a la calidad y seguridad de estos.
La regulación es el conjunto de instrumentos normativos por medio de los cuales los gobiernos establecen requisitos a los ciudadanos y las empresas, y sobre los cuales se espera un cumplimiento por parte de los actores regulados (Presidencia de la República, 2019). De acuerdo con la Organización para la Cooperación y Desarrollo Económicos (OCDE), la regulación “abarca una variedad de instrumentos mediante los cuales los gobiernos establecen requerimientos para empresas y personas” (OCDE, 2012).
En el año 2014, la OCDE hizo un estudio sobre la política normativa en Colombia e identificó que la producción normativa en el país es excesiva, genera costos adicionales y barreras para los ciudadanos y al Estado (DNP, 2019).
A partir de dicho diagnóstico, recomendó desarrollar un conjunto común y obligatorio de estándares y requisitos administrativos para diseñar mejores regulaciones por lo que se establece el Conpes 3816 de 2014 sobre “Mejora Normativa: Análisis de Impacto”, cuyo objetivo es generar las capacidades institucionales en el uso de las herramientas y buenas prácticas (DNP, 2019). Posteriormente en 2019, la Política de Mejora Normativa se incluyó en el Modelo Integrado de Planeación y Gestión (MIPG), haciéndola obligatoria en todas las instituciones públicas.
Para esto se utiliza el ciclo de gobernanza regulatoria con el fin de brindar una lectura integral de cada una de las fases, actores, herramientas e instituciones que intervienen en el proceso de diseño de una nueva norma o la modificación de una existente (DNP, 2019) y que se muestra en el gráfico.
Gráfico 8. Ciclo de gobernanza regulatoria

Fuente: (DNP, 2019).
Dentro de este ciclo, se encuentra el punto 2 sobre diseño de la regulación el cual contiene los análisis de impacto normativo, tanto ex ante como ex post, reglamentados por el Decreto número 1595 de 2015 y 1468 de 2020, los cuales solo son obligatorios para los reglamentos técnicos en Colombia.
El reglamento técnico se define como el documento en el que se establecen las características de un producto o los procesos y métodos de producción con ellas relacionadas, con inclusión de las disposiciones administrativas aplicables y cuya observancia es obligatoria. También puede incluir prescripciones en materia de terminología, símbolos, embalaje, marcado o etiquetado, aplicables a un producto, proceso o método de producción, o tratar exclusivamente de ellas.
De esta forma, el MSPS se encuentra obligado a establecer análisis de impacto normativo, tanto ex ante como ex post, para aquellos reglamentos que sean aplicables a los DM como los de buenas prácticas de manufactura (BPM) y etiquetado, entre otros. A la fecha se encuentra publicado el análisis ex post de los Decretos número 4725 de 2005 y 3770 de 2004 (Minsalud, 2023) y la problemática respecto a las BPM de DM.
Del primero se puede destacar que en torno al régimen de registros sanitarios, permisos de comercialización y vigilancia sanitaria tanto para los DM como para los reactivos de diagnóstico in vitro, se reconoce que, si bien se mantienen las problemáticas originales que dieron lugar a las intervenciones regulatorias de los Decretos número 3770 de 2004 y 4725 de 2005, las condiciones del sector han cambiado a lo largo del tiempo, por lo que estos decretos no son suficientes para responder a la problemática actual, por lo que se recomendó realizar los ajustes a esta regulación, teniendo en cuenta las oportunidades de mejora identificadas en la evaluación (Minsalud, 2023).
Alineados con este proceso, la actualización del régimen de registros sanitarios, permisos de comercialización y vigilancia sanitaria debe también responder al artículo 161 del Plan Nacional de Desarrollo 2022-2026 (Ley 2294 de 2023) que establece que se debe realizar el “fortalecimiento para agilizar las autorizaciones de los procesos de fabricación, venta e importación de medicamentos y dispositivos y tecnologías en salud”.
Ahora bien, las Buenas Prácticas de Manufactura de DM (BPM), a las que se hace mención en los Decretos número 4725 de 2005 y 3770 de 2004 y la cual actualmente se está reglamentando, comprenden los procedimientos y métodos utilizados para asegurar la calidad durante la manufactura, empaque, almacenamiento e instalación de los DM, así como, la estructura organizacional, responsabilidades, procesos y recursos para implementar los requisitos de calidad asociados con estos.
El reconocimiento de normas internacionales como la ISO:13485 sobre sistemas de gestión de calidad para la fabricación de DM (ISO 13485:2018), así como la normalización con respecto a calidad de materias primas o elementos de fabricación de los DM (IETS, 2019) no está definida. A pesar de contar con estándares como la ISO 14971:2019 (“Dispositivos médicos/productos sanitarios (MD) – Aplicación de la gestión del riesgo a los DM”), la IEC 60601-1 (“Equipos electromédicos. Parte 1: Requisitos generales para la seguridad básica y funcionamiento esencial”), la ISO 15223-1 (“Productos sanitarios. Símbolos para utilizar en las etiquetas, el etiquetado y la información a suministrar. Parte 1: Requisitos generales”), la ISO 14155 (“Investigación clínica de productos sanitarios para humanos. Buenas prácticas clínicas”), la ISO 14971 (“Productos sanitarios. Aplicación de la gestión de riesgos a los productos sanitarios”), y la UNE-EN 62353:2015 (“Equipos electromédicos. Ensayos recurrentes y ensayos después de reparación del equipo electromédicos”), a la fecha no se encuentran armonizados con la reglamentación colombiana.
Actualmente, derivado de los mencionados decretos el Invima expide un concepto técnico de condiciones sanitarias, que adolece de los requisitos planteados a nivel internacional, por lo que no es reconocido para el comercio exterior (Minsalud, 2019).
En otros temas regulatorios se encuentra que, en cuanto al mantenimiento, a la fecha no se cuenta con ningún estándar internacional con el que Colombia este alineado a pesar de que en el mundo se encuentran algunos como el AAMI EQ-56 que son las prácticas recomendadas para un programa de gestión de mantenimiento de equipos biomédicos (AAMI - Association for the Advancement of Medical Instrumentation, 2013).
Por su parte, respecto a metrología para DM, el Decreto número 1595 de 2015 y sus actos reglamentarios, se define que la calibración de los DM considerados instrumentos de medición se realiza de acuerdo con los reglamentos técnicos metrológicos y mientras se expidan estos, se hace de acuerdo con las indicaciones y periodicidad que establezca el fabricante. Menciona también que para este tipo de DM la calibración debe realizarse en un laboratorio acreditado en la norma ISO 17025, que es un estándar internacional con los requisitos generales para las competencias de los laboratorios de prueba y calibración. Para estos DM en específico existe ya una armonización con la norma internacional.
Sin embargo, para los demás equipos biomédicos a la fecha se deberán hacer las actividades de soporte técnico (mantenimiento y calibración) de acuerdo con las indicaciones del fabricante, según el Decreto número 4725 de 2005. En cuanto a lineamientos de calibración el IETS menciona que es preciso desarrollar más el cómo se debe hacer la metrología, considerando los estándares internacionales.
Se evidencia entonces que se requiere fortalecer tanto la regulación como la reglamentación técnica para los DM en Colombia.
6. Escasos lineamientos para la gestión, así como para el uso adecuado de DM.
6.1. Difusos lineamientos para la gestión de dispositivos médicos, especialmente de equipos biomédicos.
Como se mencionó en el marco conceptual, la gestión de tecnologías en salud (GTS) comprende la planificación, la evaluación de las necesidades, la selección, la adquisición, donaciones, inventario, instalación y mantenimiento de equipos médicos, formación para un uso seguro y disposición final (OMS, 2017).
En el diagnóstico realizado por el IETS una de las problemáticas identificadas está relacionada con las dificultades y barreras en la adquisición de los DM, Colombia al igual que otros países del mundo carece de un marco regulatorio sobre la gestión de los DM que permita la toma de decisiones ajustadas al perfil epidemiológico, costos, nivel complejidad, y la estructura de los servicios de salud (IETS, 2019).
Actualmente la normatividad nacional de DM contempla que el fabricante, titular e importador cuenta con obligaciones y responsabilidades relacionadas a la producción, procesamiento, envase, empaque, almacenamiento, expendio, uso, importación, exportación, comercialización y mantenimiento (Decreto número 4725 de 2005), sin embargo, carece de otros requisitos y responsabilidades específicamente definidas en temas de adquisición, venta y lo especifico para posventa y mantenimiento es escaso.
En relación con el talento humano para realizar el mantenimiento, el IETS menciona que éste debe contar con los conocimientos teóricos y prácticos en diferentes tipos de DM que les permita ostentar experiencia suficiente para intervenirlos adecuadamente en búsqueda de la seguridad del paciente y del mismo operador (IETS, 2019).
Por otro lado, en cuanto a donaciones, el IETS menciona la necesidad de contar con una Política para donaciones y puntualizó en la importancia de garantizar la donación de tecnología segura y de esta manera evitar riesgos con donaciones a poblaciones lejanas o de bajo acceso a tecnología, acorde a condiciones locales que protejan la salud de los colombianos (IETS, 2019).
Finalmente, el IETS se refiere a la urgencia de contar con reglas de juego claras para el reúso de los DM que cumplan las condiciones para este efecto (IETS, 2019).
Durante los últimos años, el MSPS como ente rector, ha realizado múltiples esfuerzos por reglamentar acciones asociadas a la gestión de DM, teniendo en cuenta el ciclo de vida en la cadena de comercialización de estas tecnologías en el país (IETS, 2019), resumida así:
- En lo relativo a la planificación y la evaluación de las necesidades, no se encuentran requisitos normativos específicos para ambas etapas.
- En relación con la selección y adquisición, la Resolución número 3100 de 2019 establece que se debe contar con información documentada de estos procesos para los DM dentro del estándar de “medicamentos, dispositivos médicos e insumos” (Resolución número 3100 de 2019).
- En cuanto a donaciones, el Decreto número 218 de 2019 regula los requisitos, el procedimiento para el ingreso y salida del territorio aduanero nacional, y las autorizaciones expedidas por el Invima relacionadas con donaciones internacionales con fines sociales y humanitarios de DM entre otras tecnologías (Decreto 218 de 2019). Así mismo, en Colombia, en el marco de la emergencia sanitaria por COVID-19, se expidió el Decreto número 697 de 2021 que a la fecha esta derogado. Sin embargo, aún no se tiene reglamentación respecto a las donaciones nacionales.
- Referente al inventario, no se encontró información más allá de los requisitos de reporte del Sistema de Información Hospitalaria (SIHO) donde se solicita información de algunos equipos biomédicos considerados DM según lo establece el Decreto número 2193 de 2004 compilado en el Decreto número 780 de 2016.
Respecto a la instalación y mantenimiento de equipos biomédicos, la Resolución número 3100 de 2019 establece que todos los prestadores de servicios de salud en Colombia deben contar con un programa de mantenimiento preventivo de los equipos biomédicos, así como con las hojas de vida donde se tengan los registros de estas actividades. Menciona también que el mantenimiento debe ser ejecutado por talento humano profesional tecnólogo o técnico en áreas relacionadas (Resolución número 3100 de 2019). También el Decreto número 1769 de 1994 regula la asignación y utilización de los recursos financieros del 5% del presupuesto total, destinados al mantenimiento de la infraestructura y de la dotación hospitalaria en los hospitales públicos, y en los privados que tengan más del 30% de sus ingresos totales provenientes de los contratos con la Nación (Decreto número 1769 de 1994).
6.2. Escasos lineamientos en el uso adecuado y seguro de DM.
Si bien hay algunos lineamientos sobre uso adecuado y seguro, aun se observa inadecuado uso de los DM en la prestación de los servicios de salud, por lo que es importante afianzar los conocimientos técnicos y fortalecer el rol consciente del personal de salud sobre el uso determinado por el fabricante, pero también, el de comunicar información privilegiada sobre riesgos potenciales, prohibiciones y contraindicaciones, que en muchas ocasiones no se evidencian por razones exclusivamente comerciales.
De igual manera, la información y capacitación sobre el uso adecuado debe estar alineada con las recomendaciones del fabricante, dado que algunos problemas de calidad pueden estar relacionados a fallas en las capacidades de los usuarios y la desinformación sobre los DM. Así mismo el IETS manifiesta que algunas veces el usuario final (paciente o cuidador) no sabe usar los DM (IETS, 2019).
Al tratar el tema de formación para un uso seguro, se encuentra que la Resolución 3100 de 2019 menciona que es indispensable contar con un programa de capacitación en el uso de DM cuando éstos lo requieran (Resolución número 3100 de 2019).
Dado la diversidad de DM para el hogar y atención en domicilio, la comunidad también debe ser informada y concientizada sobre el uso adecuado, los riesgos potenciales y contraindicaciones, situación que no se ha abordado por parte de los fabricantes, importadores y distribuidores.
OBJETIVOS
Objetivo general
Fortalecer el acceso equitativo de la población colombiana a los DM integrando los factores que inciden en su disponibilidad, calidad, continuidad, uso seguro y adecuado en la salud individual y colectiva para contribuir con los resultados en salud que se dan en el Sistema General de Seguridad Social en Salud.
Objetivos específicos
OE1. Diseñar un sistema de información de DM que integre las herramientas existentes e identifique las que se requieran para responder a las necesidades de toma de decisiones en el sector.
OE2. Establecer los lineamientos que contribuyan al fortalecimiento de la investigación, el desarrollo y la práctica clínica en materia de DM.
OE3. Robustecer la rectoría y gobernanza sobre los DM desarrollando capacidades técnicas en los actores.
OE4. Implementar estrategias para aumentar la disponibilidad y la asequibilidad de DM priorizados, que respondan al perfil epidemiológico de la población colombiana.
OE5. Fortalecer lineamientos postmercado, la regulación y la reglamentación técnica con énfasis en la promoción de la calidad y seguridad de los DM.
OE6. Desarrollar lineamientos para la gestión, así como para el uso adecuado de DM.
PLAN DE ACCIÓN
1. OE1. Diseñar un sistema de información de DM que integre las herramientas existentes e identifique las que se requieran para responder a las necesidades de toma de decisiones en el sector.
1.1. Línea de acción: Consolidar los requerimientos para el diseño del sistema de información de dispositivos médicos.
1.1.1. Entre el año 2024 y 2025, el Ministerio de Salud y Protección Social en coordinación con los actores adelantará un inventario de las fuentes de información que existen durante el ciclo de vida de los DM en el país, donde se identifique el estado, los parámetros y el nivel de madurez de sistemas de información existentes.
1.1.2. Entre el año 2024 y 2026, el Ministerio de Salud y Protección Social implementará el estándar semántico y codificación de los DM.
1.1.3. Entre el año 2024 y 2026, el Ministerio de Salud y Protección Social diseñará el sistema de información que, de forma escalonada y cibersegura, integre las herramientas existentes en el sector salud identificadas en el inventario, cree las que se requieran y que permita comparar de forma nacional e internacional la información de DM, alineados con las políticas de Gobierno Digital y el SISPRO.
1.2. Línea de acción: Analizar la información que se recolecta actualmente sobre DM.
1.2.1. Entre el año 2024 y 2026, el Ministerio de Salud y Protección Social realizará el soporte técnico y mantenimiento de los softwares asociados a DM que se alojen en esta entidad, según requerimientos y atendiendo los lineamientos de Gobierno Digital y de la Oficina de Tecnología de la Información y la Comunicación (TIC).
1.2.2. Entre el año 2024 y 2026, el Ministerio de Salud y Protección Social en coordinación con el Invima, realizará análisis de la información sobre DM que se genere con los sistemas de información existentes en la actualidad, con el fin de que esta sea de conocimiento público y que apoye la toma de decisiones.
2. OE2. Establecer los lineamientos que contribuyan al fortalecimiento de la investigación, el desarrollo y la práctica clínica en materia de dispositivos médicos.
2.1. Línea de acción: Promover la I+D+i de DM en escenarios y con los actores involucrados en las políticas de I+D+i.
2.1.1. Entre el año 2024 y 2026, el Ministerio de Salud y Protección Social en articulación con los actores involucrados en las políticas de I+D+i promoverá espacios de discusión sobre la investigación y desarrollo de DM.
2.2. Línea de acción: Establecer lineamientos sobre investigación, desarrollo y práctica clínica en DM.
2.2.1. Entre el año 2025 y 2026, el Ministerio de Salud y Protección Social en coordinación con los actores establecerá lineamientos para la investigación en salud con DM.
2.2.2. Entre el año 2025 y 2026, el Ministerio de Salud y Protección Social en articulación con el Ministerio de Comercio, Industria y Turismo, el Invima y otros actores involucrados, adecuarán los requisitos sanitarios para facilitar la importación de materia primas consideradas DM, así como los equipos para uso en investigación e innovación de DM.
3. OE3. Robustecer la rectoría y gobernanza sobre los DM desarrollando capacidades técnicas en los actores.
3.1. Línea de acción: Reforzar el conocimiento sobre gestión, evaluación y vigilancia durante el ciclo de vida de los DM, en las entidades del sector salud de carácter público a nivel nacional y territorial.
3.1.1. Entre el año 2024 y 2026, el Ministerio de Salud y Protección Social elaborará, realizará y evaluará un programa de capacitación para reforzar el conocimiento sobre gestión, evaluación y vigilancia durante el ciclo de vida de los DM en las entidades públicas del nivel central, así como en las entidades territoriales de salud.
4. OE4. Implementar estrategias para fortalecer la disponibilidad y la asequibilidad de DM priorizados, que respondan al perfil epidemiológico de la población colombiana.
4.1. Línea de acción: Mejorar la disponibilidad de los DM en el mercado.
4.1.1. Entre el año 2024 y 2026, el Ministerio de Salud y Protección Social establecerá una herramienta de monitoreo del estado de abastecimiento interoperable y de consulta pública para los actores, así como, el régimen de vigilancia al reporte que permita aumentar y gestionar la disponibilidad de DM estratégicos en el mercado del país.
4.1.2. Entre el año 2024 y 2026, el Ministerio de Salud y Protección Social en articulación con el Invima y con participación de los actores, definirá estrategias con el fin de evitar el riesgo de escasez manteniendo la calidad, seguridad, funcionamiento y desempeño de estos en los rangos que mitiguen los riesgos en salud pública.
4.2. Línea de acción: Divulgar metodologías de evaluación de tecnologías en salud según necesidades del sector.
4.2.1. Entre el año 2024 y 2026, el Ministerio de Salud y Protección Social en articulación con el IETS y los actores, establecerá, capacitará y divulgará los conceptos, metodologías, lineamientos y guías para realizar Evaluaciones de Tecnologías en Salud para DM priorizados en diferentes actores de acuerdo con el perfil epidemiológico colombiano.
4.3. Línea de acción: Contribuir al desarrollo de la política de precios de los DM.
4.3.1. Entre el año 2024 y 2026, el Ministerio de Salud y Protección Social a través de la Comisión Nacional de Precios de Medicamentos y Dispositivos Médicos, basados en la metodología para la identificación de los DM, los priorizará e incluirá, de manera progresiva, en los regímenes de la política nacional de precios.
4.4. Línea de acción: Impulsar la producción local de DM.
4.4.1. Entre el año 2024 y 2026, el Ministerio de Salud y Protección Social realizará las acciones en torno a la implementación de la política de soberanía en la producción para la seguridad sanitaria, específicamente aquellas definidas en la comisión intersectorial para el desarrollo y producción de tecnologías estratégicas en salud (CIDPTES).
4.4.2. Entre el año 2024 y 2026, el Ministerio de Salud y Protección Social en conjunto con los involucrados, articulará las acciones definidas en la Política de Reindustrialización relacionadas con las capacidades de producción local de los DM.
5. OE5. Fortalecer lineamientos postmercado, la regulación y la reglamentación técnica con énfasis en la promoción de la calidad y seguridad de los estos.
5.1. Línea de acción: Optimizar los lineamientos del sistema de vigilancia postmercado de los DM.
5.1.1. Entre el año 2025 y 2026, el Ministerio de Salud y Protección Social en articulación con el Invima, las entidades territoriales de salud y actores definirá lineamientos para promover la vigilancia proactiva, activa e intensiva dentro de los programas de vigilancia posmercado.
5.1.2. Entre el año 2024 y 2026, el Invima en articulación con el Ministerio de Salud y Protección Social, las entidades territoriales de salud y actores establecerá lineamientos para mejorar los canales de comunicación, retroalimentación y tiempos de respuesta de los programas de vigilancia postmercado, teniendo en cuenta las realidades de los territorios.
5.1.3. Entre el año 2024 y 2025, el Ministerio de Salud y Protección Social en articulación con los actores del nivel central y territorial avanzará en el diseño del modelo de inspección, vigilancia y control sanitario definido por la Resolución número 1229 de 2013 o la norma que lo modifique, adicione o sustituya, incluyendo DM.
5.2. Línea de acción: Robustecer la regulación y la reglamentación técnica de los DM entorno a la calidad y seguridad de estos.
5.2.1. Entre el año 2024 y 2025, el Ministerio de Salud y Protección Social en articulación con el Invima y la participación de los actores involucrados realizará los análisis de impacto normativo requeridos, alineados con las buenas prácticas regulatorias y las disposiciones legales aplicables vigentes.
5.2.2. Entre el año 2024 y 2025, el Ministerio de Salud y Protección Social en articulación con el Invima y la participación de los actores involucrados, avanzará en la reglamentación técnica de las Buenas Prácticas de Manufactura de los DM en serie.
5.2.3. Entre el año 2025 y 2026, el Ministerio de Salud y Protección Social en articulación con los actores, establecerá una propuesta de lineamientos para el mantenimiento y el uso de la metrología de equipos biomédicos articulada con el Subsistema Nacional de Calidad, donde se incluya revisión de los lineamientos internacionales existentes en el tema.
5.2.4. Entre el año 2024 y 2026, el Ministerio de Salud y Protección Social en articulación con los actores actualizará el régimen de registros sanitarios, permisos de comercialización y vigilancia sanitaria de DM para uso humano.
6. OE6. Desarrollar lineamientos para la gestión, así como para el uso adecuado de DM.
6.1. Línea de acción: Establecer lineamientos para la gestión de dispositivos médicos, especialmente de equipos biomédicos.
6.1.1. Entre el año 2024 y 2025, el Ministerio de Salud y Protección Social en articulación con los actores y agentes, definirá lineamientos para la gestión de DM.
6.1.2. Entre el año 2024 y 2026, el Ministerio de Salud y Protección Social con participación de las entidades territoriales de salud, los prestadores de servicios de salud y otros actores e involucrados, crearán estrategias para la divulgación de los conocimientos en mejores prácticas de gestión de DM a nivel hospitalario.
6.1.3. Entre el año 2024 y 2026, el Ministerio de Salud y Protección Social en articulación con los actores, definirán e implementarán lineamientos para la reprocesamiento de dispositivos y elementos reutilizables (DMER) en los prestadores de servicios de salud (PSS) y avanzarán en la construcción de lineamientos para la reprocesamiento y remanufactura de dispositivos médicos de único uso (DMUU).
6.1.4. Entre el año 2024 y 2026, el Ministerio de Salud y Protección Social en articulación con los actores definirá lineamientos sobre disposición final tanto hospitalaria como comunitaria de los DM especialmente para DM implantables, así como para equipos biomédicos incluyendo la mitigación de los impactos en la sostenibilidad ambiental.
6.2. Línea de acción: Promover el uso adecuado de DM de acuerdo con los lineamientos del fabricante y las necesidades derivadas del perfil epidemiológico.
6.2.1. Entre el año 2025 y 2026, el Ministerio de Salud y Protección Social en articulación con los actores y agentes desarrollará estrategias para impulsar el uso adecuado e idóneo de los DM que sean priorizados, de acuerdo con los lineamientos del fabricante, en los prestadores de servicios de salud, los pacientes y los cuidadores.
6.2.2. Entre el año 2025 y 2026, el Ministerio de Salud y Protección Social en articulación con el Invima establecerá lineamientos para la publicidad de DM.
SEGUIMIENTO
La Dirección de Medicamentos y Tecnologías en Salud y la Oficina Asesora de Planeación y Estudios Sectoriales y del Ministerio de Salud y Protección Social realizarán un seguimiento anual, en el periodo comprendido entre 2024 y 2026, a la ejecución de las acciones propuestas para el cumplimiento del objetivo de la presente política, presentando informes de avance anuales y un informe de cierre final posterior al año 2026.
El Plan de Acción y Seguimiento (PAS) establecido para la ejecución de la Política de Dispositivos Médicos será publicado por la Dirección de Medicamentos y Tecnologías en Salud al igual que los informes de avances anuales y de cierre final.
FINANCIAMIENTO
Para el cumplimiento de los objetivos de esta política, las entidades del sector salud involucradas en su implementación gestionarán y priorizarán, en el marco de sus competencias y de acuerdo con el Marco de Gasto de Mediano Plazo, los recursos para la financiación de las acciones que son propuestas.
BIBLIOGRAFÍA
AAMI - Association for the Advancement of Medical Instrumentation. (2013). AAMI EQ56 - Recommended practice for a medical equipment management program.
Aguilar, V. L. (2006). Gobernanza y gestión Pública. Obtenido de https:// marcelagonzalezduarte.files.wordpress.com/2018/01/aguilar-villanueva-gobernanza-y-gestion-publica.pdf.
Burmester, H. (1997). Reflexiones sobre los programas hospitalarios de garantía de la calidad. Rev Panam Salud Pública.
CEPAL. (2020). La salud como desafío productivo y tecnológico. Obtenido de https:// www.cepal.org/sites/default/files/publication/files/46534/S2000782_es.pdf.
CEPAL. (2021). Primeras lecciones y desafíos de la pandemia de COVID-19 para los países del SICA.
Cheng, M. (2003). Medical Device Regulation: Global overview and guiding principles. Geneve: World Health Organization.
Decreto número 1099 de 2022. (s. f.). Por el cual se crea la Comisión Intersectorial para el Desarrollo y Producción de Tecnologías Estratégicas en Salud (CIDPTES).
Decreto número 1769 de 1994. (s. f.). Por el cual se reglamenta el artículo 90 del Decreto número 1298 de 1984.
Decreto número 218 de 2019. (s. f.). Por el cual se regula las donaciones internacionales de productos de uso humano con fines sociales y humanitarios y se dictan otras disposiciones.
Decreto número 3770 de 2004. (12 de noviembre de 2004). Por el cual se reglamentan el régimen de registros sanitarios y la vigilancia sanitaria de los reactivos de diagnóstico in vitro para exámenes de especímenes de origen humano. Obtenido de https://www. minsalud.gov.co/Normatividad_Nuevo/DECRETO%203770%20DE%202004.PDF.
Decreto número 4107 de 2011. (s. f.). Por el cual se determinan los objetivos y la estructura del Ministerio de Salud y Protección Social y se integra el Sector Administrativo de Salud y Protección Social.
Decreto número 4725 de 2005. (s. f.). Por el cual se reglamenta el régimen de registros sanitarios, permiso de comercialización y vigilancia sanitaria de los dispositivos médicos para uso humano. Obtenido de https://www.alcaldiabogota.gov.co/sisjur/normas/Norma1. jsp?i=18697&dt=S.
Decreto número 697 de 2021. (s. f.). Por el cual se establecen los requisitos sanitarios que se tendrán en cuenta en la donación de medicamentos de síntesis química, biológicos. radiofármacos, productos fitoterapéuticos, gases medicinales, medicamentos homeopáticos, dispositivos médicos, equipos biomédicos, reactivos de diagnóstico in vitro, cosméticos y productos de higiene doméstica y absorbentes de higiene personal, y materias primas, para atender la pandemia por la COVID-19.
DNP. (2019). Política de Mejora Normativa para Modernizar el Estado. Obtenido de https://colaboracion.dnp.gov.co/CDT/ModernizacionEstado/EReI/GME_PolItica_ Mejora_Normativa.pdf.
Escobar, N. (2000). La Innovación tecnológica. Editorial Medisan.
Fernández, M. D. (1998). La calidad y la innovación tecnológica en la biotecnología aplicada a la salud. Obtenido de http://revistadyo.es/index.php/dyo/article.
FDA. (2023). Software as a Medical Device (SaMD). Página web https://www.fda. gov/medical-devices/digital-health-center-excellence/software-medical-device-samd consultada el 23 de agosto de 2023.
FIFARMA. (2019). Obtenido de https://fifarma.org/wp-content/uploads/2019/10/ FIFARMA-Healthcare-Sustainability-Working-Document-20190903-STC-Spanish.pdf.
García-Vega, Óscar Armando. (2015). Presentación “Sistema nacional de vigilancia de dispositivos médicos” en el XII Encuentro Internacional de Farmacovigilancia de las Américas. Obtenido de: https://ciemto.medicinaudea.co/system/comfy/cms/ files/files/000/000/260/original/8-Sistema_nacional_de_vigilancia_de_dispositivos_ biome%CC%81dicos.pdf.
Gilmore. C., N. H. (1996). Gerencia de calidad. Washington D. C: OPS.
IETS. (2014). Manual metodológico para la elaboración de evaluaciones de efectividad, seguridad y validez diagnóstica de tecnologías en salud. Obtenido de https:// www.minsalud.gov.co/sites/rid/Lists/BibliotecaDigital/RIDE/INEC/IETS/manual-metodologico-elaboracion-de-evaluaciones-de-efectividad.pdf.
IETS. (2019). Documento técnico de diagnóstico del sector de los dispositivos médicos. Bogotá.
IMDRF. (2019) IMDRF MDCE WG/N57FINAL:2019 (formerly GHTF/SG5/ N3:2010) Clinical Investigation. Obtenido de https://www.imdrf.org/sites/default/files/ docs/imdrf/final/technical/imdrf-tech-191010-mdce-n57.pdf.
ISO 13485:2018. (s. f.). Productos sanitarios. Sistemas de gestión de la calidad. Requisitos para fines reglamentarios.
Kaur M et al. (2005). How to Manage' Series for Healthcare Technology. Guide 1: How to Organize a System of Healthcare Technology Management. Ziken International (Health Partners International).
Kirigia J, K. D. (2011). International Archives of Medicine. Obtenido de http://www. intarchmed.com/content/4/1/11. Consultado el 20 de febrero de 2013).
Laudon, L. &. (2004). Obtenido de http://cotana.informatica.edu.bo/downloads/ld- Sistemas_de_informacion_gerencial_14%20edicion.pdf.
Ley 1438 de 2011. (s. f.). Por medio de la cual se reforma el Sistema General de Seguridad Social en Salud y se dictan otras disposiciones. Obtenido de https://www.suin-juriscol. gov.co/viewDocument.asp?ruta=Leyes/1680431#:~:text=Es%20obligaci%C3%B3n%20 de%20la%20familia,su%20desarrollo%20arm%C3%B3nico%20e%20integral.
Ley 842 de 2003. (s. f.). Por la cual se modifica la reglamentación del ejercicio de la ingeniería, de sus profesiones afines y de sus profesiones auxiliares, se adopta el Código de Ética Profesional y se dictan otras disposiciones.
Borderline and Classification Working Group - Medical Device Coordination Group (2022). Manual on borderline and classification for medical devices under Regulation (EU) 2017/745 on medical devices and Regulation (EU) 2017/746 on in vitro diagnostic medical devices. Version 2.
Mincit. (20 de 2 de 2023). Página web Ministerio de Comercio, Industria y Turismo. Obtenido de https://www.mincit.gov.co/prensa/noticias/industria/politica-reindustrializacion-presentacion-a-medios.
Minsalud. (2015). Mejorar la seguridad en la utilización de medicamentos. Obtenido de https://www.minsalud.gov.co/sites/rid/Lists/BibliotecaDigital/RIDE/DE/CA/seguridad-en-la-utilizacion-de-medicamentos.pdf
Minsalud. (2019). Línea base sobre estado de los dispositivos médicos en Colombia. Bogotá.
Minsalud. (2021). Documento preliminar de AIN de BPM de DM. Bogotá.
Minsalud. (2023). Informe evaluación expost Decretos 3770 de 2004 y 4725 de 2005. Bogotá. Consultado en https://www.minsalud.gov.co/Anexos_Normatividad_ Nuevo/20232400028190300004%20%281%29.pdf.
Minsalud. (2023b). Árbol de problemas en el marco del Análisis de Impacto Normativo (AIN) de Investigación Clínica con Dispositivos Médicos (DM) y Reactivos de Diagnóstico In Vitro (RDIV). Bogotá
Molina, D., Botero, S., & Giraldo, G. (2016). Investigación clínica y ensayos clínicos.
Molina-Marín, A. N.-D. (2012). Rectoría y gobernanza en salud pública. Obtenido de https://scielosp.org/pdf/rsap/2013.v15n1/44-55/es.
Monter, H. A. (2008). ¿Qué sabe usted acerca de …eficacia y efectividad de los fármacos? Obtenido de https://www.redalyc.org/pdf/579/57939108.pdf.
OCDE. (2 de 11 de 2012). Recomendación del Consejo sobre Política y Gobernanza Regulatoria.
OCDE. (2015). Manual de Frascati. Obtenido de Guía para la recopilación y presentación de información sobre la investigación y el desarrollo experimental: https:// minciencias.gov.co/sites/default/files/manual_de_frascati_web_0_1.pdf).
OCDE. (2018). Oslo Manual 2018: Guidelines for Collecting, Reporting and Using Data on Innovation, (4th Edition ed.). OCDE Publishing.
OMS. (2007). Resolución WHA 60.29. Ginebra.
OMS. (2012). Dispositivos meídicos: la gestioín de la discordancia: un resultado del proyecto sobre dispositivos meídicos prioritarios. Genéve: Organizacioín Mundial de la Salud. Obtenido de https://apps.who.int/iris/handle/10665/44868.
OMS. (2012). Formulación de políticas sobre dispositivos médicos. Obtenido de http://apps.who.int/iris/bitstream/handle/10665/44832/9789243501635_spa.pdf;jsessioni d=DC39707277502CF14CDB604195602044?sequence=1.
OMS. (2017). Global Atlas of medical devices. Obtenido de https://www.who.int/ publications/i/item/9789241512312
OMS. (2017). Modelo Global de Marco Regulatorio de la OMS para Dispositivos Médicos, incluidos los dispositivos médicos para diagnóstico in vitro. Ginebra.
OMS. (2020). Guideline on Country Pharmaceutical Pricing Policies.
OPS. (2012). 28.a CONFERENCIA SANITARIA PANAMERICANA. Obtenido de https://iris.paho.org/bitstream/handle/10665.2/3684/CSP28.R9-s. pdf?sequence=4&isAllowed=y.
OPS. (2023). Equidad en Salud. Obtenido de https://www.paho.org/es/temas/equidad-salud.
OPS, U. (2007). LA FUNCIÓN RECTORA de la AUTORIDAD SANITARIA NACIONAL. Obtenido de https://www.paho.org/hq/dmdocuments/2010/Rectoria_ASN_ Lecciones_Aprendidas_Rectoria_Conduccion.pdf.
OPS/OMS. (Mayo de 2021). Evaluación de tecnologías de salud.
Presidencia de la República. (Diciembre de 2019). Política de Mejora Normativa. Comité de Mejora Normativa Consejo para la Gestión y Desempeño Institucional. Versión 1.
RAE. (2021). Obtenido de https://dle.rae.es/seguro?m=form
Resolución número 1411 de 2022. (s. f.). Por la cual se adopta la Política de Soberanía en la producción para la Seguridad.
Resolución número 2366 de 2023. (s. f.). Por la cual se establecen los servicios y tecnologías de salud financiados con recursos de la Unidad de Pago por Capitación (UPC).
Resolución número 3100 de 2019. (s. f.). Por la cual se definen los procedimientos y condiciones de inscripción de los prestadores de servicios de salud y de habilitación de los servicios de salud y se adopta el Manual de Inscripción de Prestadores y Habilitación de Servicios de Salud.
Resolución número 4816 de 2008. (s. f.). Por la cual se reglamenta el Programa Nacional de Tecnovigilancia.
Rivas, L. V. (2006). Ingeniería Clínica y Gestión de Tecnología en Salud: Avances y Propuestas. Obtenido de http://its.uvm.edu/PUCP_CENGETS/LIBRO-CENGETS-NOV2006.pdf.
Suplemento, O. (2008). La calidad de la atención en salud. Obtenido de https://www. medigraphic.com/pdfs/oral/ora-2008/oras081a.pdf).
Unión Medical. (s. f.) Escasez de dispositivos médicos por pandemia. Obtenido de https://um.com.co/blog/escasez-de-dispositivos-medicos-por-pandemia/#proveedores.
Vega-Pérez et al. (2017). Obtenido de http://www.scielo.org.co/pdf/rori/v21n1/0121- 3709-rori-21-01-00064.pdf
Voury y De Geyndt. (2008). La calidad de la atención en salud. Obtenido de https:// www.medigraphic.com/pdfs/oral/ora-2008/oras081a.pdf.
Las notas de vigencia, concordancias, notas del editor, forma de presentación y disposición de la compilación están protegidas por las normas sobre derecho de autor. En relación con estos valores jurídicos agregados, se encuentra prohibido por la normativa vigente su aprovechamiento en publicaciones similares y con fines comerciales, incluidas -pero no únicamente- la copia, adaptación, transformación, reproducción, utilización y divulgación masiva, así como todo otro uso prohibido expresamente por la normativa sobre derechos de autor, que sea contrario a la normativa sobre promoción de la competencia o que requiera autorización expresa y escrita de los autores y/o de los titulares de los derechos de autor. En caso de duda o solicitud de autorización puede comunicarse al teléfono 617-0729 en Bogotá, extensión 101. El ingreso a la página supone la aceptación sobre las normas de uso de la información aquí contenida.