|
|
|
CIRCULAR 14 DE 2022
(julio 25)
Diario Oficial No. 52.116 de 4 de agosto de 2022
COMISIÓN NACIONAL DE PRECIOS DE MEDICAMENTOS Y DISPOSITIVOS MÉDICOS
<NOTA DE VIGENCIA: Circular derogada por el artículo 46 de la Circular 15 de 2023>
Por la cual se establecen disposiciones generales para el seguimiento del comportamiento del mercado de dispositivos médicos, se incorporan y mantienen algunos de estos productos en el régimen de libertad vigilada, se conserva el estent coronario medicado en el régimen de control directo, se estructura el sistema de reporte de información de precios de dispositivos médicos y se dictan otras disposiciones.
LA COMISIÓN NACIONAL DE PRECIOS DE MEDICAMENTOS Y DISPOSITIVOS MÉDICOS,
en ejercicio de sus facultades legales, en especial de las conferidas por los artículos 245 de la Ley 100 de 1993, 87 de la Ley 1438 de 2011, y 3o. del Decreto número 1071 de 2012, y
CONSIDERANDO:
Que por su parte, en el literal j) del artículo 5o. de la Ley Estatutaria 1751 de 2015, establece que el Estado es responsable de respetar, proteger y garantizar el goce efectivo del derecho fundamental a la salud; para ello deberá, entre otros, intervenir el mercado de medicamentos y dispositivos médicos en la salud, con el fin de optimizar su utilización, evitar las inequidades en el acceso, asegurar la calidad de los mismos, o en general, cuando pueda derivarse una grave afectación de la prestación del servicio.
Que el artículo 60 de la Ley 81 de 1988 contempla las modalidades bajo las cuales se basa el ejercicio de la política de precios de diversos productos, entre los cuales se incluyen los medicamentos y dispositivos médicos, estableciendo los regímenes de control directo, libertad regulada y libertad vigilada.
Que el artículo 90 de la Ley 1438 de 2011, estableció que el Gobierno nacional de Colombia, en aras de garantizar la competencia efectiva para la producción, venta, comercialización y distribución de medicamentos y dispositivos médicos, podrá adelantar las gestiones necesarias para que la población disponga de estos productos con buena calidad y a precios accesibles.
Que, a través de la Comisión Nacional de Precios de Medicamentos y Dispositivos Médicos (CNPMDM), se han definido lineamientos atinentes a sistemas de información, su funcionamiento, formas de reporte de precio de estos productos y actualizaciones frente a la inclusión o exclusión de dispositivos en los diferentes regímenes de control y vigilancia.
Que, por otra parte, la política de precios de dispositivos médicos tiene por objeto hacer compatibles las necesidades de salud pública con los principios de competencia, comercio y viabilidad de la inversión para producir beneficios en salud que justifiquen sus costos, con una aplicación clara, simple y práctica para los usuarios.
Que, en virtud de lo anterior, la Comisión tiene la facultad para hacer inclusión de dispositivos médicos al régimen de control directo de precios, al de libertad vigilada o al de libertada regulada, utilizando para el efecto herramientas como: investigaciones, estudios de sistemas empleados en otros países, mesas de trabajo con los participantes y usuarios de los servicios de salud, en procura de beneficios para estos y un mercado consecuente.
Que los países de referencia internacional utilizados en el cálculo del precio máximo de venta han sido seleccionados de conformidad con los criterios de: i) integración comercial, ii) proximidad geográfica con Colombia, iii) similitud en el grado de intervención económica general o política de regulación de precios de dispositivos médicos o iii) pertenencia a la Organización para la Cooperación y el Desarrollo Económicos (OCDE), conforme a la disponibilidad y calidad de la información. En total son 17 países y 37 bases de datos públicas y de libre acceso, algunas de ellas son online y otras son documentos descargables en PDF o Excel.
Que si bien todos los precios de los dispositivos médicos impactan la sostenibilidad del Sistema General de Seguridad Social en Salud (SGSSS) y podrían estar en libertad vigilada, al no ser esto posible por las dificultades para codificar y estandarizar los dispositivos médicos en Colombia, se procedió a realizar una priorización que obedece a una metodología que se encuentra publicada en la página web del Ministerio de Salud y Protección Social en el enlace: https://www.minsalud.gov.co/sites/rid/Lists/ BibliotecaDigital/RIDE/VS/MET/metodologia-analisis-nuevos-dm-22dic2021.pdf
Que esta metodología analiza la base de datos de recobros por concepto de dispositivos médicos de la Administradora de los Recursos del Sistema General de Seguridad Social en Salud (ADRES) para los años 2019 y 2020 identificando aquellos cuyo impacto en el monto total del valor aprobado en recobros es alto. Esto se logra tras clasificar, aplicar el principio de Pareto y posteriormente seleccionar los dispositivos médicos que se deban considerar para su inclusión en los regímenes de control de precios.
Que como consecuencia de la aplicación de esta metodología se incluyen cuatro (4) nuevos mercados relevantes de dispositivos médicos al régimen de libertad vigilada los cuales son implantes auditivos de conducción ósea, implante para córnea, implante coclear y dispositivos médicos implantables para terapia cardiaca de alto voltaje.
Que, la Comisión Nacional de Precios de Medicamentos y Dispositivos Médicos (CNPDM), expidió la Circular número 02 de 2017, modificada parcialmente por la Circular 05 de 2018, y en su Anexo Técnico 1 se establece la forma y metodología para realizar el reporte de precios de dispositivos médicos en la Plataforma de Intercambio de Información (PISIS), en donde se incluyen datos relevantes de dispositivos médicos.
Que a la fecha los obligados a reportar envían los archivos planos con la información de los precios de venta de dispositivos médicos sujetos a los regímenes de libertad vigilada y control directo al Ministerio de Salud y Protección Social y lo hacen a través de la Plataforma de Intercambio de Información (PISIS) del Sistema Integral de Información de la Protección Social (SISPRO). Sin embargo, este medio no cuenta con una denominación en la reglamentación por lo que se hace necesario asignarle el nombre de Sistema de Información de Precios de Dispositivos Médicos (SISDIS) y establecer los objetivos y forma de operación para dar claridad a los regulados.
Que el reporte actual solo cuenta con la información de los precios de venta de dispositivos médicos sujetos a los regímenes de libertad vigilada y control directo, evidenciando que esta estructura dificulta la trazabilidad de las transacciones y no necesariamente refleja la configuración de la cadena de suministro, por lo que, la Comisión encuentra necesario ampliar esta estructura de reporte para tener mejor información del mercado de dispositivos médicos, que permita guiar las políticas en salud y reducir las asimetrías en la información.
Que, para esto, se hace necesario definir los requerimientos del SISDIS e incluir información sobre el tipo de transacción, la unidad de reporte, los precios de compra y de recobro de dispositivos médicos, así como los roles que los obligados a reportar tienen dentro de la cadena de comercialización.
Que, con lo anterior el SISDIS busca convertirse en una herramienta idónea para el análisis y monitoreo de precios, y así mejorar las capacidades regulatorias y de vigilancia que permita detectar las distorsiones del mercado.
Que la CNPMDM pretende promover la transparencia en el sector de la salud y espera que esta modificación en el anexo técnico del SISDIS facilite el acceso a información de precios de dispositivos médicos.
Que por otro lado, desde el año 2015 y hasta la fecha, la Comisión ha expedido diversos actos que regulan la materia de la siguiente manera: Circular 01 de 2015, a través de la cual se interviene en los mercados de dispositivos médicos, se establece un régimen de libertad vigilada para los mismos y se someten unos estents coronarios a control directo, surtiendo modificaciones por medio de las Circulares 2 del mismo año; Circular 02 de 2016, a través de la cual se introducen ajustes por variación de precios a los estents coronarios; Circular 02 de 2017, modificada por la Circular 05 de 2018, que incorpora al Régimen de Libertad Vigilada los dispositivos médicos anticonceptivos, se sustituye el Anexo Técnico número 1, adoptado con la Circular 1 y la Circular 13 de 2021, que ajusta precio de los estents coronarios.
Que, en el año 2019 el Grupo Técnico Asesor (GTA) de la Comisión realizó mesas de trabajo para conocer la percepción de los actores que intervienen en la implementación de la regulación de precios de dispositivos médicos. En dichas mesas, los interesados refirieron que, al existir múltiples circulares sobre la materia, se genera confusión entre los actores al aplicarlas y desconocimiento de estos en la forma en como la autoridad competente realizaba la inspección, vigilancia y control.
Que, en virtud de lo descrito, se considera pertinente proferir un acto administrativo que incluya las temáticas dispuestas en las circulares previamente citadas con el objeto de otorgar mayor claridad y optimizar su aplicación.
Que los temas que se van a regular a través de este acto, no limitan el número o la variedad de las empresas en el mercado ni la capacidad de estas para competir, razón por la cual no resulta necesario solicitar concepto de abogacía de la competencia ante la Superintendencia de Industria y Comercio.
Que el articulado completo del proyecto de la presente Circular fue sometido a consulta pública entre el 22 de noviembre de 2021 y 14 de diciembre de 2021, recibiendo comentarios de la ciudadanía que se analizaron y constituyen el fundamento de algunas de las decisiones adoptadas en la presente Circular.
En mérito de lo expuesto,
RESUELVE:
OBJETO Y DEFINICIONES.
ARTÍCULO 1o. OBJETO. <Circular derogada por el artículo 46 de la Circular 15 de 2023> La presente circular tiene por objeto establecer disposiciones generales para el seguimiento del comportamiento del mercado de dispositivos médicos; incorporar y mantener algunos de estos productos en el régimen de libertad vigilada; conservar el estent coronario medicado en el régimen de control directo y estructurar el sistema de reporte de información de precios de dispositivos médicos.
ARTÍCULO 2o. ÁMBITO DE APLICACIÓN. <Circular derogada por el artículo 46 de la Circular 15 de 2023> La presente Circular aplica a:
2.1 Fabricantes, importadores, comercializadores y titulares de registro sanitario de dispositivos médicos
2.2. Prestadores de servicios de salud en todos los regímenes, incluyendo los regímenes Especiales y de Excepción.
ARTÍCULO 3o. DEFINICIONES. <Circular derogada por el artículo 46 de la Circular 15 de 2023> Para efectos de la presente circular, además de las definiciones contenidas en el Decreto número 4725 de 2005, serán aplicables las siguientes:
Anillo intracorneal: Banda circular implantable y abierta en un extremo, para aplanar la curvatura anterior de la córnea sin afectar al eje visual, a fin de corregir la miopía leve o moderada. Se inserta en la córnea a través de un canal intraestromal creado con un bisturí de diamante o láser. Es de plástico y tiene un diámetro externo e interno de aproximadamente 8 y 6,8 mm, respectivamente.
Anticonceptivos de barrera: Dispositivo anticonceptivo en forma de vaina que crea una barrera física y es destinado a evitar la fecundación o a evitar la transmisión de enfermedades de transmisión sexual (ETS). Puede tener textura, sabor o contener lubricantes. Se trata de un dispositivo de un solo uso.
Anticonceptivos invasivos: Dispositivo anticonceptivo diseñado para introducirse (se implanta sin cirugía) dentro de la cavidad uterina, cerca del fondo, y evitar la fecundación. Normalmente conocido como DIU, lo coloca un profesional sanitario cualificado usando dispositivos que habitualmente se incluyen (p. ej., un introductor). Se trata de un dispositivo de un solo uso.
Canal institucional: corresponde al canal donde se realizan las transacciones de compra y venta de dispositivos médicos con recursos públicos. Hacen parte del canal institucional los agentes que intervienen en la cadena de compra, venta y recobro de dispositivos médicos con recursos públicos, que incluye a los vendedores y compradores de regímenes especiales.
Canal comercial: corresponde al canal donde se realizan las transacciones de compra y venta de dispositivos médicos con recursos que no son públicos.
Conjunto de productos relevantes para la regulación de precios por su sustituibilidad terapéutica o funcional: Corresponde al conjunto de dispositivos médicos para los que se reconoce sustituibilidad terapéutica y/o funcional, considerando la evidencia científica disponible o su uso. Estos pueden corresponder a más de un mercado relevante por existir evidencia de baja sustituibilidad económica entre subconjuntos de estos dispositivos médicos.
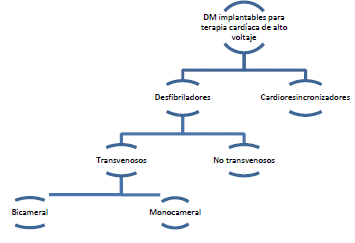
Dispositivos médicos implantables para terapia cardiaca de alto voltaje - desfibrilador transvenoso bicameral: Generador de impulsos implantable, con un sistema de reconocimiento del ritmo cardíaco, que permite analizar un electrocardiograma y suministra un impulso eléctrico para desfibrilar el corazón (restablecer su ritmo normal) o disminuir la frecuencia cardíaca, así como para estimular el corazón en caso de bradicardia. Cuenta con derivaciones que se colocan dentro o sobre dos cavidades cardíacas homolaterales (aurícula y ventrículo) para monitorizar los parámetros electrocardiográficos y brindar de forma automática el impulso eléctrico. Incluye baterías internas que proporcionan la energía necesaria para suministrar los impulsos, y se conoce habitualmente como cardioversor-desfibrilador implantable automático.
Dispositivos médicos implantables para terapia cardiaca de alto voltaje-resincronizadores: Dispositivo implantable estéril alimentado por baterías que consta de un generador de impulsos de electroestimulación cardíaca, cerrado herméticamente y un generador de impulsos de desfibrilación integrado con derivaciones colocadas en el ventrículo derecho, en una vena coronaria sobre el ventrículo izquierdo y, con frecuencia, en la aurícula derecha (tricameral). Además de las funciones de desfibrilación y marcapasos convencionales, el aparato suministra tratamiento de resincronización cardíaca mediante estimulación eléctrica biventricular, con el fin de sincronizar las contracciones de los ventrículos derecho e izquierdo, de forma que aumente la eficacia del bombeo sanguíneo, lo que permite tratar síntomas de insuficiencia cardíaca (p. ej., respiración entrecortada, fatiga rápida) y problemas graves relacionados con el ritmo cardíaco (desfibrilador TRSC).
Dispositivos médicos implantables para terapia cardiaca de alto voltaje-desfibrilador transvenoso monocameral: Generador de impulsos implantable, con un sistema de reconocimiento del ritmo cardíaco, que permite analizar un electrocardiograma y suministra un impulso eléctrico para desfibrilar el corazón (restablecer su ritmo normal) o disminuir la frecuencia cardíaca. Cuenta con derivaciones que se colocan dentro o sobre una cavidad cardíaca (normalmente, un ventrículo) para monitorizar los parámetros electrocardiográficos y brindar de forma automática el impulso eléctrico que permite tratar la fibrilación ventricular o la taquicardia. Incluye baterías internas que proporcionan la energía necesaria para suministrar las descargas, y se conoce habitualmente como cardioversor-desfibrilador implantable automático.
Dispositivos médicos para implante coclear: Conjunto de dispositivos (implante coclear [IC]) alimentado por baterías diseñado para la restauración parcial de la sensación auditiva en una persona con sordera profunda. Consta de una matriz electrodos interna que se implanta en la cóclea y se conecta a un estimulador receptor implantado cerca del oído, un procesador de sonido y micrófono colocado externamente, una unidad de batería, el cable de la bobina y la propia bobina (transmisor). El procesador de sonido (transductor) convierte las ondas sonoras en señales eléctricas y las envía a la bobina (transmisor) a través del cable correspondiente. La bobina transmite las señales al estimulador receptor y finalmente a la cóclea a través de la matriz de electrodos. Pueden estar incluidos el instrumental de implantación especializado.
Estent Coronario Convencional: Dispositivo endovascular coronario implantable de forma permanente, cuyo fin es mejorar el espacio luminal del vaso sin liberar medicamento alguno.
Estent Coronario Medicado: Dispositivo endovascular coronario implantable de forma permanente, cuyo fin es mejorar el espacio luminal del vaso liberando algún medicamento.
Implante auditivo de conducción ósea: Dispositivo acústico externo, alimentado eléctricamente, destinado a compensar la discapacidad auditiva mediante la transmisión de vibraciones, desde ondas sonoras traducidas, a través del cráneo hasta el oído interno. Por lo general, incorpora un micrófono, amplificador y vibrador conectados a un imán externo que está acoplado magnéticamente a un imán que no vibra implantado en la cabeza para su retención; puede incluir dispositivos de posicionamiento y protección. El micrófono recibe ondas sonoras y las convierte en señales eléctricas enviadas al vibrador que transmite vibraciones a los oídos internos para escuchar. Este dispositivo se usa generalmente para tratar la discapacidad auditiva debido a patologías obstructivas del oído medio y / o externo. Comprende el audífono de conducción ósea: implante magnético y el sistema de implante de oído medio parcialmente implantable.
Mercado relevante para efectos de regulación de precios: Corresponde a un grupo de dispositivos médicos para las cuales existe evidencia científica sobre similitud en las características del producto y su sustituibilidad terapéutica y/o funcional, así como la información de precios de que no existen diferencias significativas asociadas a las características terapéuticas del producto o de su uso. Para la conformación de estos mercados relevantes las diferencias en precios del producto de las asociaciones de marca no serán un criterio relevante para el análisis de sustitución económica, ni de segmentación adicional.
Punto de la cadena regulado: Es el punto de la cadena de suministro en la que se fija el precio máximo de venta.
Reporte en cero: Todo fabricante, importador o comercializador de los dispositivos médicos incluidos en los regímenes de libertad vigilada, libertad regulada o control directo que tengan registro sanitario y no realice ventas durante el período de reporte deberá en todo caso realizar el reporte en ceros.
Segmento anular intracorneal: Arco implantable para aplanar la curvatura anterior corneal sin alterar el eje visual, con el fin de corregir determinados trastornos visuales relacionados con la córnea. Es un arco de plástico de unos 150 grados que se suministra por pares, se introduce en la córnea a través de un canal intraestromal realizado con un bisturí de diamantes o láser, y puede implantarse lejos de la incisión radial para reducir la posibilidad de que surjan complicaciones. El tamaño oscila entre 0,25 y 0,35 milímetros. Se utiliza para corregir la miopía leve o moderada y tratar a los pacientes que presentan astigmatismo irregular provocado por queratocono, especialmente aquellos que no pueden beneficiarse del uso de lentes de contacto o gafas.
Transacción primaria: Corresponde a la primera venta en el territorio nacional del dispositivo médico objeto de reporte, por parte del actor que fabrica o importa a otro actor, incluyendo los regímenes especiales y de excepción. Igualmente, corresponde a la primera compra en el territorio nacional del dispositivo médico objeto de reporte al actor que lo fabrica o lo importa.
Transacción secundaria: Corresponde a las ventas posteriores a la transacción primaria del dispositivo médico objeto de reporte que va a ser suministrado o usado. Entendiéndose esta operación de venta proveniente de un actor que no lo fabrica ni lo importa a otro actor, incluyendo los regímenes especiales y de excepción. Igualmente, corresponde a la compra del dispositivo médico objeto de reporte que va a ser suministrado o usado. Entendiéndose esta operación de compra a un actor que no lo fabrica ni lo importa, incluyendo los regímenes especiales y de excepción.
Transacción final: Corresponde a la venta del dispositivo médico objeto de reporte que haya sido vendido con fines de ser suministrado a un paciente, incluyendo los regímenes especiales y de excepción. Corresponde al recobro/cobro del dispositivo médico objeto de reporte que haya sido suministrado a un paciente, incluyendo los regímenes especiales y de excepción. Igualmente, es la compra del dispositivo médico objeto de reporte que haya sido comprado con fines de ser suministrado a un paciente.
Se exceptúan del reporte aquellas operaciones de comodato, alquiler o arriendo de dispositivos médicos.
DISPOSICIONES GENERALES.
ARTÍCULO 4o. FUENTES DE INFORMACIÓN. <Circular derogada por el artículo 46 de la Circular 15 de 2023> La Comisión iniciará la inclusión sistemática de los dispositivos médicos al régimen de libertad vigilada. Adicionalmente y durante la implementación de este régimen, así como para la aplicación del régimen de control directo o de libertad regulada, utilizará las fuentes de información disponibles entre otras. La Comisión preferirá el uso de información pública y siempre revelará la fuente de información. La Comisión hará pública la información utilizada siempre y cuando esta no sea de carácter confidencial en los términos establecidos en la legislación colombiana.
PARÁGRAFO. La Secretaría Técnica de la Comisión podrá solicitar a cualquier miembro de la cadena de distribución, información necesaria para las labores de control y vigilancia de precios de dispositivos médicos.
ARTÍCULO 5o. PERÍODO DE REFERENCIA. <Circular derogada por el artículo 46 de la Circular 15 de 2023> Los datos utilizados serán los contemplados en las fuentes de información relacionadas en el artículo 4o. de este acto, durante el período comprendido entre el 1o. de enero al 31 de diciembre del año inmediatamente anterior a la expedición del acto administrativo que regule los precios de los dispositivos médicos
ARTÍCULO 6o. IDENTIFICACIÓN DE MERCADOS RELEVANTES. <Circular derogada por el artículo 46 de la Circular 15 de 2023> La Comisión a través de la Secretaría Técnica realizará revisión y análisis de la información sobre el comportamiento de los precios, así como de los recobros de los dispositivos médicos, una vez por año, para identificar la necesidad de establecer nuevos mercados relevantes que sean sujetos a regulación de precios.
PARÁGRAFO. Para la actualización de los precios que han sido regulados, se realizará una nueva referenciación internacional.
ARTÍCULO 7o. ETAPAS DE ANÁLISIS DE LOS MERCADOS DE DISPOSITIVOS MÉDICOS A INGRESAR A LIBERTAD VIGILADA. <Circular derogada por el artículo 46 de la Circular 15 de 2023> La metodología de análisis de los dispositivos médicos a ingresar a libertad vigilada consta de las siguientes etapas:
7.1. Identificación de aquellos dispositivos médicos que impactan la sostenibilidad del Sistema General de Seguridad Social en Salud (SGSSS), considerando entre otros, aquellos con mayor participación en el valor total recobrado a la Administradora de los Recursos del Sistema General de Seguridad Social en Salud (ADRES), estos últimos se priorizarán seleccionando aquellos dispositivos con mayor participación en el valor de los recobros, que conjuntamente acumulen entre el 50% y el 80% del valor total recobrado.
7.2. Definición del conjunto de productos relevantes para su inclusión al régimen de libertad vigilada.
7.3. Análisis a nivel nacional de los mercados relevantes; donde se consideran, entre otros, los siguientes criterios:
a) El total de oferentes y la concentración de mercado.
b) La pertenencia o no al Plan de Beneficios en Salud (PBS).
ARTÍCULO 8o. DETERMINACIÓN DEL CONJUNTO DE PRODUCTOS RELEVANTES PARA LA REGULACIÓN DE PRECIOS POR SU SUSTITUIBILIDAD TERAPÉUTICA Y/O FUNCIONAL. <Circular derogada por el artículo 46 de la Circular 15 de 2023> Para la regulación de precios, se entenderán como un conjunto de productos relevantes aquellos que sean sustituibles de forma terapéutica y/o funcional. Para esto se tendrá en cuenta:
8.1. Agrupaciones realizadas por nomencladores internacionales de dispositivos médicos o el estándar semántico adoptado por Colombia para dispositivos médicos de uso humano, cuando se haya establecido.
8.2. En caso de requerirse, se complementará la información con la revisión documental de las guías, manuales y recomendaciones de uso dadas por el fabricante del dispositivo médico.
8.3. Cuando se considere necesario, la Secretaría Técnica de la Comisión determinará las asociaciones profesionales y otros expertos del sector de los dispositivos médicos que se estén definiendo y realizará las acciones para consultar sus recomendaciones sobre el conjunto de productos relevantes, en cuyo caso las respuestas harán parte de las actas que evidencien las recomendaciones.
DISPOSITIVOS MÉDICOS ENDOVASCULARES CORONARIOS.
ARTÍCULO 9o. CONSERVACIÓN DE DISPOSITIVOS MÉDICOS EN EL RÉGIMEN DE LIBERTAD VIGILADA. <Circular derogada por el artículo 46 de la Circular 15 de 2023> Todos los dispositivos médicos endovasculares coronarios con registro sanitario otorgado por el Instituto Nacional de Vigilancia de Medicamentos y Alimentos (Invima) que se comercialicen a nivel nacional, se mantienen en el régimen de libertad vigilada.
ARTÍCULO 10. DETERMINACIÓN DEL CONJUNTO DE PRODUCTOS RELEVANTES POR SUSTITUIBILIDAD TERAPÉUTICA Y/O FUNCIONAL. <Circular derogada por el artículo 46 de la Circular 15 de 2023> Los dispositivos médicos endovasculares coronarios se definen como un conjunto de productos relevantes.
ARTÍCULO 11. CONJUNTO DE DISPOSITIVOS MÉDICOS ENDOVASCULARES CORONARIOS. <Circular derogada por el artículo 46 de la Circular 15 de 2023> El conjunto de dispositivos médicos endovasculares coronarios se clasifica de la siguiente manera:
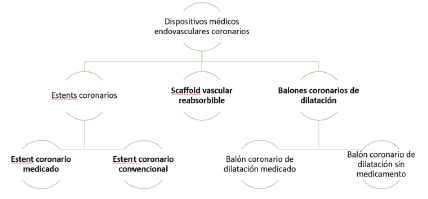
ARTÍCULO 12. MERCADOS RELEVANTES OBJETO DE ANÁLISIS. <Circular derogada por el artículo 46 de la Circular 15 de 2023> Los mercados relevantes objeto de análisis son los “estent coronarios”, el cual a su vez se divide en dos mercados relevantes: 1) “estent coronario convencional” y 2) “estent coronario medicado”.
ARTÍCULO 13. RÉGIMEN DE CONTROL DIRECTO. <Circular derogada por el artículo 46 de la Circular 15 de 2023> Se mantienen en el régimen de control directo todos los estent coronarios medicados que se comercializan a nivel nacional.
ARTÍCULO 14. PUNTO DE LA CADENA REGULADO. El precio se regula para las transacciones finales institucionales. Esto implica que el precio de regulación corresponde al precio máximo de venta que se puede cobrar en cualquier transacción a lo largo de la cadena. Los prestadores no podrán aumentar al precio regulado en ningún porcentaje en sus transacciones con los proveedores y/o pagadores. El precio regulado corresponde también al valor máximo de recobro a la Administradora de los Recursos del Sistema General de Seguridad Social en Salud (ADRES).
ARTÍCULO 15. ESTABLECIMIENTO DEL PRECIO DE REFERENCIA NACIONAL (PRN) PARA LOS ESTENT CORONARIOS MEDICADOS. <Circular derogada por el artículo 46 de la Circular 15 de 2023> El PRN es el precio expresado en unidad de medida, característico de un mercado relevante en Colombia. Se calcula como el promedio ponderado de los precios unitarios expresados en unidad de medida por las cantidades vendidas, utilizando la información de las transacciones finales institucionales reportadas al SISDIS para el período de referencia de que trata el artículo 5o. de la presente Circular, teniendo en cuenta los titulares de registro sanitario de los estent coronarios medicados que hagan parte del mercado relevante.
ARTÍCULO 16. APLICACIÓN DEL FACTOR DE AJUSTE PARA LOS ESTENT CORONARIOS. <Circular derogada por el artículo 46 de la Circular 15 de 2023> A los precios obtenidos de bases de datos de países de referencia que tengan información de precios en el punto ex fábrica, se aplicará un factor de ajuste de 9%, para hacerlos comparables con el punto de la cadena regulado.
ARTÍCULO 17. ESTABLECIMIENTO DEL PRECIO DE REFERENCIA INTERNACIONAL (PRI) PARA EL ESTENT CORONARIO MEDICADO. <Circular derogada por el artículo 46 de la Circular 15 de 2023> El PRI corresponde al precio expresado en unidad de medida de acuerdo con el percentil 25 calculado a partir de los datos de precios de los países de referencia, cuando estén disponibles, y se establece de la siguiente manera:
17.1. Consulta de los precios internacionales: se consultará el precio del estent coronario medicado en las fuentes de información de los países de referencia definidos en el artículo 19 de la presente Circular. De existir más de un precio para un país, se calculará el promedio simple de estos precios para determinar un precio por país.
17.2. Cálculo del Precio de Referencia Internacional (PRI): se calculará con base en el percentil 25 de los precios disponibles en los países de referencia y por unidad de medida para cada mercado relevante.
ARTÍCULO 18. TEMPORALIDAD DE LOS DATOS DE PRECIOS A OBSERVAR. <Circular derogada por el artículo 46 de la Circular 15 de 2023> Los precios, tanto nacionales como internacionales, utilizados para el análisis de los diferentes mercados de dispositivos médicos, serán aquellos correspondientes a la información más reciente disponible en las bases de datos consultadas. Esta información deberá corresponder con el período de referencia del que trata el artículo 5o. de la presente circular.
ARTÍCULO 19. PAÍSES DE REFERENCIA INTERNACIONAL. <Circular derogada por el artículo 46 de la Circular 15 de 2023> Son los países a partir de los cuales la Comisión obtiene la información de los precios de dispositivos médicos, los cuales corresponden a los siguientes: Alemania, Argentina, Australia, Brasil, Canadá, Chile, Ecuador, España, Estados Unidos, Francia, Grecia, Italia, México, Panamá, Perú, Portugal, Reino Unido y Uruguay.
ARTÍCULO 20. NÚMERO DE PAÍSES PARA REFERENCIAR. <Circular derogada por el artículo 46 de la Circular 15 de 2023> La referenciación internacional deberá realizarse, por lo menos, con información de tres países de los mencionados en el artículo 19.
ARTÍCULO 21. TASA DE CAMBIO. <Circular derogada por el artículo 46 de la Circular 15 de 2023> Se utiliza para comparar los precios de dispositivos médicos obtenidos en los países de referencia internacional con los precios en el mercado nacional. En el marco de la presente metodología, para los precios expresados en una unidad monetaria distinta a pesos colombianos (COP), se tomará el promedio de venta y compra de la divisa publicada de manera diaria por el Banco de la República. Para establecer la tasa de cambio se calcula el promedio móvil con un rezago de 20 días de la tasa de cambio nominal para cada día del período de referencia, para luego establecer el promedio simple de los valores calculados de este periodo como la tasa de cambio a utilizar en la ficha de regulación.
ARTÍCULO 22. PRECIO MÁXIMO DE VENTA (PMV). <Circular derogada por el artículo 46 de la Circular 15 de 2023> El Precio Máximo de Venta (PMV) para los mercados relevantes sujetos a la aplicación de régimen de control directo de precios de dispositivos médicos corresponderáí al menor valor entre el PRN y el PRI.
ARTÍCULO 23. APLICACIÓN DEL PRECIO MÁXIMO DE VENTA PARA LOS ESTENTS CORONARIOS MEDICADOS. <Circular derogada por el artículo 46 de la Circular 15 de 2023> El Precio Máximo de Venta de los estents coronarios medicados es de $2.858.499,75 COP.
PARÁGRAFO. Para el caso del estent coronario medicado los ajustes metodológicos que la presente circular incorpora, serán aplicados únicamente para las futuras regulaciones de este dispositivo, por lo tanto, difiere de la metodología que se utilizó para establecer el precio máximo de venta presentado en la Circular 13 de 2021 la cual se unifica en el presente acto administrativo.
ARTÍCULO 24. PROHIBICIÓN DE INCREMENTAR EL PRECIO MÁXIMO DE VENTA POR INTERMEDIACIÓN. <Circular derogada por el artículo 46 de la Circular 15 de 2023> El Precio Máximo de Venta del estent coronario medicado, será el máximo permitido para realizar operaciones en la cadena de comercialización y ningún actor de la cadena que realice operaciones en las transacciones institucionales y comerciales, podrán sobrepasar el precio regulado.
DISPOSITIVOS MÉDICOS ANTICONCEPTIVOS.
ARTÍCULO 25. CONSERVACIÓN DE DISPOSITIVOS MÉDICOS EN EL RÉGIMEN DE LIBERTAD VIGILADA. <Circular derogada por el artículo 46 de la Circular 15 de 2023> Todos los dispositivos anticonceptivos con registro sanitario otorgado por el Instituto Nacional de Vigilancia de Medicamentos y Alimentos (Invima) que se comercialicen a nivel nacional, se mantienen en el régimen de libertad vigilada.
ARTÍCULO 26. CONJUNTO DE DISPOSITIVOS MÉDICOS ANTICONCEPTIVOS. <Circular derogada por el artículo 46 de la Circular 15 de 2023> El conjunto de dispositivos médicos anticonceptivos se clasifica de la siguiente manera:
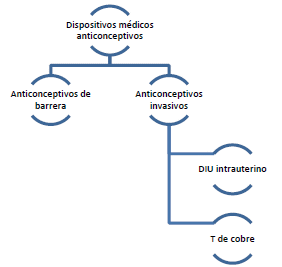
ARTÍCULO 27. EXCEPCIONES. <Circular derogada por el artículo 46 de la Circular 15 de 2023> Si el anticonceptivo es usado en prestadores de servicios de salud para exámenes diagnósticos, no se aplica el régimen de libertad vigilada.
INCORPORACIÓN DE NUEVOS DISPOSITIVOS MÉDICOS AL RÉGIMEN DE LIBERTAD VIGILADA.
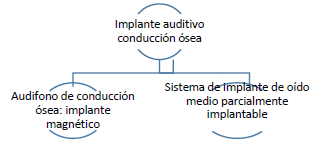
ARTÍCULO 28. INCORPORACIÓN DE LOS IMPLANTES AUDITIVOS DE CONDUCCIÓN ÓSEA A RÉGIMEN DE LIBERTAD VIGILADA. <Circular derogada por el artículo 46 de la Circular 15 de 2023> Incorpórense al régimen de libertad vigilada, el conjunto de dispositivos médicos implantes auditivos de conducción ósea con registro sanitario otorgado por el Invima que se comercialicen a nivel nacional que se clasifican de la siguiente manera:
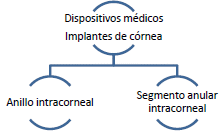
ARTÍCULO 29. INCORPORACIÓN DE DISPOSITIVOS MÉDICOS DE IMPLANTE PARA CÓRNEA A RÉGIMEN DE LIBERTAD VIGILADA. <Circular derogada por el artículo 46 de la Circular 15 de 2023> Incorpórense al régimen de libertad vigilada, el conjunto de dispositivos médicos para implante de córnea con registro sanitario otorgado por el INVIMA que se comercialicen a nivel nacional que se clasifican de la siguiente manera:
ARTÍCULO 30. INCORPORACIÓN DE DISPOSITIVOS MÉDICOS PARA IMPLANTE COCLEAR A RÉGIMEN DE LIBERTAD VIGILADA. <Circular derogada por el artículo 46 de la Circular 15 de 2023> Incorpórense al régimen de libertad vigilada los implantes cocleares con registro sanitario otorgado por el Invima que se comercialicen a nivel nacional.
ARTÍCULO 31. INCORPORACIÓN DE DISPOSITIVOS MÉDICOS IMPLANTABLES PARA TERAPIA CARDIACA DE ALTO VOLTAJE A RÉGIMEN DE LIBERTAD VIGILADA. <Circular derogada por el artículo 46 de la Circular 15 de 2023> Incorpórense al régimen de libertad vigilada, el conjunto de dispositivos médicos de implantables para terapia cardiaca de alto voltaje con registro sanitario otorgado por el Invima que se comercialicen a nivel nacional y que se clasifican de la siguiente manera:
SISTEMA DE INFORMACIÓN DE PRECIOS DE DISPOSITIVOS MÉDICOS (SISDIS).
ARTÍCULO 32. SISTEMA DE INFORMACIÓN DE PRECIOS DE DISPOSITIVOS MÉDICOS (SISDIS). <Circular derogada por el artículo 46 de la Circular 15 de 2023> Conjunto de componentes interrelacionados que trabajan juntos para recopilar, procesar, almacenar y difundir información sobre precios de los dispositivos médicos incluidos en los regímenes de libertad vigilada, libertad regulada o control directo que se comercialicen en el territorio nacional. El reporte en SISDIS se realiza en la Plataforma de Intercambio de Información (PISIS) del Sistema Integral de Información de la Protección Social (SISPRO).
Este sistema tendrá como objetivo:
32.1. Normalizar el registro, almacenamiento, flujo, transferencia y disposición de la información para la regulación del mercado de dispositivos médicos en toda la cadena de producción, importación y distribución.
32.2. Contribuir a la disminución de las asimetrías de información existentes en el sector, a través de la disposición y uso de información uniforme, integrada y de calidad.
32.3 Facilitar el acceso a la información no reservada sobre precios de dispositivos médicos a los actores del Sistema General de Salud y al público en general, en armonía con las políticas del Gobierno en esta materia.
ARTÍCULO 33. OBLIGADOS A REPORTAR EN SISDIS. <Circular derogada por el artículo 46 de la Circular 15 de 2023> Los fabricantes o importadores de los dispositivos médicos que cuenten con registro sanitario expedido por el Invima e incorporados al régimen de libertad vigilada, regulada y control directo y las personas naturales o jurídicas que comercialicen o suministren dichos dispositivos, serán responsables de efectuar el reporte de precios, a través de Sistema de Información de Precios de Dispositivos Médicos (SISDIS). Los establecimientos farmacéuticos minoristas correspondientes a farmacias/droguerías y droguerías del canal comercial no están obligados a realizar el presente reporte.
PARÁGRAFO. Los obligados a reportar de que trata este artículo, en ningún caso deben incluir las siguientes operaciones que se realicen al detal con personas naturales: i) compra, ii) venta, iii) cobro o recobro iv) suministro, entendido como la dispensación o entrega del dispositivo del médico al paciente o su uso asociado a un procedimiento en salud.
ARTÍCULO 34. INFORMACIÓN A REPORTAR EN SISDIS. <Circular derogada por el artículo 46 de la Circular 15 de 2023> Los actores establecidos en el artículo 33, deberán reportar de cada dispositivo médico incorporado al régimen de libertad vigilada, regulada o control directo, la información que se encuentra detallada en el Anexo Técnico.
PARÁGRAFO 1o. Los fabricantes o importadores de los dispositivos médicos que cuenten con registro sanitario expedido por el Invima y que hayan sido incorporados a los regímenes de libertad vigilada, regulada y control directo que no realicen ventas durante el periodo de reporte, deberán realizar el reporte en ceros.
PARÁGRAFO 2o. En ningún caso se deben reportar muestras médicas o donaciones.
ARTÍCULO 35. PERÍODO DEL REPORTE. <Circular derogada por el artículo 46 de la Circular 15 de 2023> El reporte de información se deberá realizar trimestralmente discriminando las operaciones y transacciones para cada mes en los plazos máximos previstos en el siguiente calendario:
| Fecha de Corte de la Información a reportar | Meses que deben reportarse | Plazo para enviar el archivo plano | |
| Desde | Hasta (Fecha máxima de reporte) | ||
| 31 de marzo | Enero, febrero y marzo del respectivo año. | Primer día hábil de abril | Último día hábil de abril |
| 30 de junio | Abril, mayo, y junio del respectivo año. | Primer día hábil de julio | Último día hábil de julio |
| 30 de septiembre | Julio, agosto y septiembre del respectivo año. | Primer día hábil de octubre | Último día hábil de octubre |
| 31 de diciembre | Octubre, noviembre y diciembre del respectivo año. | Primer día hábil de enero del año siguiente | 15 de febrero del año siguiente o en caso de no ser un día hábil, el día hábil siguiente |
PARÁGRAFO. Cargue extemporáneo. Se habilitará dos veces el sistema de información de precios de dispositivos médicos -SISDIS para que los obligados a reportar, que no lo hayan hecho dentro de los períodos previamente establecidos, realicen cargues extemporáneos. La primera fecha será del 1 al 21 de mayo y la segunda del 1 al 21 de noviembre, de cada vigencia.
ARTÍCULO 36. RESPONSABILIDADES FRENTE A LA INFORMACIÓN CONTENIDA EN EL SISDIS. <Circular derogada por el artículo 46 de la Circular 15 de 2023> Los actores del Sistema General de Seguridad Social en Salud que participen en el flujo y consolidación de la información serán responsables de la privacidad, seguridad, confidencialidad y veracidad de la información suministrada y sobre los datos de los cuales se tiene acceso.
PARÁGRAFO. Para garantizar la seguridad y veracidad de la información reportada, los actores reportantes deberán enviar los archivos firmados digitalmente, garantizando su confidencialidad, integridad y no repudio. Para firmar digitalmente los archivos, se debe usar un certificado digital emitido por una entidad certificadora abierta aprobada por la entidad competente.
ARTÍCULO 37. SOPORTE Y ASISTENCIA TÉCNICA. <Circular derogada por el artículo 46 de la Circular 15 de 2023> Con el propósito de brindar asesoría y asistencia técnica, el Ministerio de Salud y Protección Social dispone de la Mesa de Ayuda de PISIS, cuyo detalle de operación se especifica en el Anexo Técnico de la presente circular.
ARTÍCULO 38. DESHABILITACIÓN DEL ANEXO TÉCNICO DE SISDIS EN LA PLATAFORMA PISIS. <Circular derogada por el artículo 46 de la Circular 15 de 2023> Después de haber sido habilitado el anexo técnico de SISDIS en la plataforma PISIS, este solo podrá ser deshabilitado, cuando los obligados a reportar en SISDIS lo soliciten y adjunten la justificación y la evidencia que la soporte.
DISPOSICIONES FINALES.
ARTÍCULO 39. PLAN PILOTO DEL SISTEMA DE INFORMACIÓN DE PRECIOS DE DISPOSITIVOS MÉDICOS - SISDIS. <Circular derogada por el artículo 46 de la Circular 15 de 2023> Todos los actores obligados a reportar en el marco de la presente Circular, contarán con un periodo de seis (6) meses de prueba en la plataforma contados a partir del primer día hábil de enero de 2023 y hasta el 30 de junio del año 2023, periodo en el cual deberán reportar de forma pedagógica el último trimestre del año 2022 y el primer trimestre del año 2023 de conformidad con lo establecido en el Anexo Técnico adoptado mediante este acto y con el acompañamiento de la Secretaría Técnica de la Comisión.
ARTÍCULO 40. SOLICITUD DE INFORMACIÓN. <Circular derogada por el artículo 46 de la Circular 15 de 2023> La Secretaría Técnica de la Comisión podrá solicitar en cualquier momento y a cualquier miembro de la cadena de distribución, la información necesaria para efectos de monitoreo y control de precios de dispositivos médicos.
ARTÍCULO 41. SANCIONES POR EL INCUMPLIMIENTO DE LO ESTABLECIDO EN LA PRESENTE CIRCULAR. <Circular derogada por el artículo 46 de la Circular 15 de 2023> La inobservancia de las disposiciones sobre los reportes de precios de dispositivos médicos y de precio máximo de venta, constituyen una violación de las disposiciones sobre protección al consumidor y su incumplimiento estará sujeto a las multas y demás sanciones administrativas contempladas en la Ley 1438 de 2011.
ARTÍCULO 42. TRANSITORIEDAD. <Circular derogada por el artículo 46 de la Circular 15 de 2023> Se tendrán en cuenta las siguientes disposiciones transitorias:
42.1. Los artículos del 32 al 38 de la presente Circular, entrarán a regir el 1o. de julio de 2023 y a partir de esa fecha, el envío de los reportes, se hará conforme a lo establecido en el Anexo Técnico de la presente Circular; mientras los artículos en mención entran a regir, el reporte de precios de dispositivos médicos seguirá realizándose de acuerdo con lo establecido en los artículos 3o., 4o., 5o., 6o., 7o., 8o., 9o. y Anexo Técnico de la Circular número 02 de 2017.
42.2. El primer reporte, que se realice posterior al 1o. de julio de 2023, corresponderá a la información del segundo trimestre del año 2023 (abril, mayo, junio). Este reporte deberá ser realizado en el plazo establecido en el artículo 35 de la presente Circular.
ARTÍCULO 43. VIGENCIA Y DEROGATORIA. <Circular derogada por el artículo 46 de la Circular 15 de 2023> La presente Circular rige a partir de la fecha de su publicación en el Diario Oficial y las disposiciones aquí contenidas, serán exigibles a partir del 1o. de octubre de 2022, fecha a partir de la cual se derogan, las Circulares números 01 y 02 de 2015, 02 de 2016, 05 de 2018, 13 de 2021 y los artículos 1o. y 2o. de la Circular número 02 de 2017. Los artículos 3o., 4o., 5o., 6o., 7o., 8o., 9o. y Anexo Técnico de la Circular número 02 de 2017, quedarán derogados el 1o. de julio de 2023.
Publíquese y cúmplase.
Dada en Bogotá, D. C., a 25 de julio de 2022.
El Ministro de Salud y Protección Social,
Fernando Ruiz Gómez.
La Ministra de Comercio, Industria y Turismo,
María Ximena Lombana Villalba.
La Delegada del Presidente de la República,
Clara Elena Parra Beltrán.
ESPECIFICACIONES DEL ARCHIVO DE REPORTE DE PRECIOS DE VENTA DE DISPOSITIVOS MÉDICOS INCORPORADOS AL RÉGIMEN DE LIBERTAD VIGILADA, REGULADA Y CONTROL DIRECTO.
<Circular derogada por el artículo 46 de la Circular 15 de 2023>
Los actores obligados a reportar definidos en la presente circular deben enviar al Ministerio de Salud y Protección Social los archivos planos con la información de los precios de dispositivos médicos. Para este anexo técnico se definen tres capítulos:
1. Estructura y especificación de los archivos.
2. Características de los archivos planos.
3. Plataforma para el envío de archivos.
1. Estructura y especificación de los archivos.
1. 1. Estructura y especificación del nombre del archivo.
El nombre de los archivos de la información de precios de dispositivos médicos debe ser enviada por los actores enunciados en el artículo 33, y debe cumplir con el siguiente estándar:
| Componente del Nombre de Archivo | Valores Permitidos o Formato | Descripción | Longitud Fija | Requerido |
| Módulo de información | DIS | Identificador del módulo de información Ejemplo: REC: Reportes de Control | 3 | SÍ |
| Tipo de Fuente | NNN | Fuente de la InformaciónEjemplo:125: Fuente ICBF | 3 | SÍ |
| Tema de información | BBBB | Información de precios de dispositivos médicos | 4 | SÍ |
| Fecha de Corte | AAAAMMDD | Fecha de corte de la información reportada. Es el último día calendario del período de información reportada. No se debe utilizar ningún tipo de separador. Fecha Válida: 20200331 | 8 | SÍ |
| Tipo de identificación del actor que reporta | ZZ | Tipo de identificación del actor que reporta la información. - Si es un actor diferente a entidad territorial: se debe especificar NI correspondiente al tipo de identificación Número de Identificación Tributaria - NIT. - Si corresponde a una Dirección Territorial del orden Departamental: se debe especificar DE- Si corresponde a una Dirección Territorial del orden Distrital: se debe especificar Dl- Si corresponde a una Dirección Territorial del orden Municipal: se debe especificar MU | 2 | SÍ |
| Número de identificación del actor que reporta | 9999999999 | Número de identificación del actor que envía los archivos, de acuerdo con el tipo de identificación del campo anterior:- Número de NIT sin incluir el dígito de verificación para tipo de Identificación NI. Para el caso de las entidades territoriales, en este campo va el código DANE. - Si es DE en este campo va el código DANE del departamento. Ejemplo 000000000025Si es DI en este campo va el código DANE del Distrito. Ejemplo 000000011001- Si es MU en este campo va el código DANE del municipio. Ejemplo 000000025001Sin excepción, se debe usar el carácter CERO de relleno a la izquierda si es necesario para completar el tamaño del campo. Ejemplo: 000860999123Nota: cuando los obligados a reportar tengan sucursales, el reporte de cada una de ellas debe reunirse y consolidarse en un solo archivo. Tal consolidado debe enviarse por un único actor, bajo un solo NIT. | 12 | SÍ |
| Extensión del archivo | .TXT | Extensión del archivo plano. Para el archivo plano.txt | 4 | SÍ |
Nombre del archivo
| Tipo de Archivo | Nombre de Archivo | Longitud |
| Reporte de información de precios de dispositivos médicos | DISNNNBBBBAAAAMMDDZZ999999999999.txt | 36 |
1. 2. Contenido del archivo.
A continuación, consideraciones para el contenido de los archivos:
El archivo de la información de precios de dispositivos médicos está compuesto por un único registro de control (Registro Tipo 1) utilizado para identificar el actor fuente de la información y varios tipos de registros de detalle numerados a partir del Registro Tipo 2, que contienen la información solicitada, así:
| Registro | Descripción | Reporte |
| Tipo 1 | Registro de control | Obligatorio |
| Tipo 2 | Registro de detalle de precios de compra, venta y recobro | Obligatorio |
Cada registro está conformado por campos, los cuales se separan por pipe (|)
1. 2. 1. Registro tipo 1 - Registro de control.
Es obligatorio. Es el primer registro que debe aparecer en los archivos que sean enviados.
| No. | Nombre del campo | Longitud máxima del campo | Tipo | Valores permitidos | Requerido |
| 0 | Tipo de registro | 1 | N | 1: valor que significa que el registro es de control | SÍ |
| No. | Nombre del campo | Longitud máxima del campo | Tipo | Valores permitidos | Requerido |
| 1 | Tipo de identificación del actor que reporta | 2 | A | ZZ- Si es un actor diferente a entidad territorial: se debe especificar NI correspondiente al tipo de identificación NIT- En el caso de las entidades territoriales, se especifica de la siguiente manera:- MU: Municipio- DE: Departamento- DI: Distrito | SÍ |
| 2 | Número de identificación del actor que reporta | 12 | N | Número de identificación del actor que envía los archivos, de acuerdo con el tipo de identificación del campo anterior:- Número de NIT sin incluir el dígito de verificación para tipo de Identificación NI. - Para el caso de las entidades territoriales, en este campo va el código DANE. - Si es DE en este campo va el código DANE del departamento. Ejemplo: 25- Si es DI en este campo va el código DANE del Distrito. Ejemplo: 11001- Si es MU en este campo va el código DANE del municipio. Ejemplo: 25001 | SÍ |
| 3 | Fecha inicial del período de la información reportada | 10 | F | En formato AAAA-MM-DD. Debe corresponder a la fecha de inicio del período de información reportada. Ejemplo fecha válida: 2021-01-01 | SÍ |
| 4 | Fecha final del período de la información reportada | 10 | F | En formato AAAA-MM-DD. Debe corresponder a la fecha final del periodo de información reportada y debe concordar con la fecha de corte del nombre del archivo. Último día calendario del mes o periodo que se está reportando. Ejemplo fecha válida: 2021-03-31 | SÍ |
| 5 | Número total de registros de detalle contenidos en el archivo | 10 | N | Debe corresponder a la cantidad de registros tipo 2, contenidos en el archivo. Si existen más registros será la suma de todos los registros. | SÍ |
1.2.2. Registro tipo 2 - Registro de detalle de precios de venta, compra y recobro de dispositivos médicos.
Mediante el registro tipo 2, los actores reportan el detalle de la información de las operaciones (compra, venta y recobro/cobro) mensuales, discriminando por transacción para cada dispositivo médico.
| No. | Nombre del campo | Longitud máxima del campo | Tipo | Valores permitidos | Requerido |
| 0 | Tipo de registro | 1 | N | 2: valor que significa que el registro es de detalle | SÍ |
| 1 | Consecutivo de registro | 10 | N | Número consecutivo de registros de detalle dentro del archivo. Inicia en 1 para el primer registro de detalle y va incrementando de 1 en 1, hasta el final del archivo. | SÍ |
| 2 | Código de habilitación | 10 | N | Cada prestador de servicios de salud inscrito en el Registro Especial de Prestadores de Servicios de Salud (REPS), deberá unificar la información de precios a nivel de la entidad territorial, incluyendo cada una de las sedes de su jurisdicción asociadas al mismo código de prestador entregado por REPS. Luego de consolidar por entidad territorial, se deberá allegar esta información para el reporte bajo un único NIT en un solo archivo plano, reportando para cada código de habilitación una línea diferente. Se deberá incluir, en este campo, los primeros 10 dígitos del código de habilitación del prestador de servicios de salud, según se le haya asignado en el REPS. En caso de no tener código de habilitación, según normatividad vigente, se debe diligenciar este campo con cero (0). Los códigos de habilitación y el NIT del reportante deben coincidir con REPS. | SÍ |
| 3 | Mes de la información a reportar | 2 | N | Corresponde al mes en el cual se efectuó la transacción del dispositivo médico de acuerdo con la factura. Debe corresponder a un mes válido perteneciente al trimestre que se está reportando. Ejemplo: 03 (marzo), 04 (abril). | SÍ |
| 4 | Canal | 3 | A | Dato usado para discriminar las ventas de los mecanismos por canal de distribución institucional y comercial. Los valores permitidos son:INS: institucionalCOM: comercial. | SÍ |
| No. | Nombre del Campo | Longitud Máxima del Campo | Tipo | Valores permitidos | Requerido |
| 5 | Rol del actor reportante frente a la operación de reporte del dispositivo médico | 1 | N | Tipo de Actor que reporta la información. Puede tomar el valor de: 1- Actor, que fabrica o importa el dispositivo médico a reportar. 2- Actor que participa en la cadena de comercialización pero que no elabora, ni importa, ni suministra o ni usa el dispositivo médico a reportar. 3- Actor que suministra o usa el dispositivo médico. En el caso de haber reportado el actor 3, se debe tener un código de habilitación. | SÍ |
| 6 | Tipo de operación | 2 | A | Corresponde al tipo de operación realizada por el actor reportante. Si el valor reportado en el campo 4, corresponde a “1”, este campo puede tomar el valor de: VN: operación de venta RC: operación de cobro/recobroSi el valor reportado en el campo 4, corresponde a “2” o “3”, este campo puede tomar el valor de: VN: operación de venta CM: operación de compra RC: operación de cobro/recobro | SÍ |
| 7 | Tipo de transacción | 2 | N | Corresponde al tipo de transacción realizada por el actor reportante. Este campo puede tomar el valor de: 01- Transacción primaria 02- Transacción secundaria 03- Transacción Final | SÍ |
| 8 | Identificador del dispositivo médico | 8 | N | Es un código identificador para los Dispositivos Médicos sujetos a reporte, según su presentación comercial y referencia. Ver la tabla de referencia “Dispositivos Médicos Libertad Vigilada” en la página web. sispro.gov.co, donde encontrará los códigos asignados. En caso de que algún dispositivo médico incorporado al régimen de libertad vigilada no se encuentre en la tabla de referencia “Dispositivos Médicos Libertad Vigilada”, se debe enviar la alerta con el número de expediente o el registro sanitario al correo sisdis@minsalud.gov. co | SÍ |
| 9 | Unidad en la que se factura el dispositivo médico en la operación reportada | 1 | A | Hace referencia a la unidad en la cual se van a reportar los siguientes campos:(10) Total de unidades (12) Precio unitario mínimo (13) Precio unitario máximo En el caso que el reporte se haga por presentación comercial, el valor permitido en este campo es:A: Presentación comercial En el caso que el reporte no se haga por presentación comercial, el valor permitido es:B: Unidad mínima de Dispensación o uso. Ejemplo: Un (1) estent coronario, un (1) condón. Se aclara que la Unidad en la que se debe realizar el reporte, debe coincidir con la Unidad en la que se factura el dispositivo médico para cada una de las operaciones reportadas al SISDIS. | SÍ |
| 10 | Total de unidades de reporte en el mes para la operación y la transacción. | 7 | N | Cantidad total de unidades en la que se factura el dispositivo médico en el mes, para la transacción y operación que se está reportando. | SÍ |
| 11 | Valor total facturado del dispositivo médico durante el mes, para la operación y transacción de reporte. | 16 | N | Suma en pesos colombianos del valor total facturado del dispositivo médico durante el mes, para la operación y transacción reportada. El campo permite hasta dos cifras decimales, utilizando el separador de punto (. ). | SÍ |
| 12 | Precio unitario mínimo del dispositivo médico durante el mes, para la operación y transacción de reporte. | 14 | N | Precio mínimo en pesos colombianos, de la unidad en la que se factura el dispositivo médico, de aquellos facturados durante el mes, para la operación y transacción reportada. El campo permite hasta dos cifras decimales, utilizando el separador de punto (. ). Este campo debe ser igual o menor que el campo 13. Debe tenerse en cuenta que el reporte se realiza por unidad en la que se factura el dispositivo médico definida en el campo 9. | SÍ |
| No. | Nombre del campo | Longitud máxima del campo | Tipo | Valores Permitidos | Requerido |
| 13 | Precio unitario máximo del dispositivo médico durante el mes, para la operación y transacción de reporte. | 14 | N | Precio máximo en pesos colombianos, de la unidad en la que se factura el dispositivo médico, de aquellos facturados durante el mes, para la operación y transacción reportada. El campo permite hasta dos cifras decimales, utilizando el separador de punto (.). Este campo debe ser igual o mayor que el campo 12. En el caso de haber en el mes una sola operación y transacción del reporte, este campo deberá ir en cero (0) y se deben diligenciar los campos 11 y 12. Debe tenerse en cuenta que el reporte se realiza por unidad en la que se factura el dispositivo médico definida en el campo 9. | SÍ |
| 14 | NIT del actor con quien se realizó la operación y transacción de reporte, al precio mínimo. | 12 | N | Número de Identificación Tributaria (NIT), sin incluir el código de verificación, del actor con el cual se realizó la operación y transacción para el mes de reporte en la factura de menor valor. | |
| 15 | Número de factura o documento equivalente del precio mínimo unitario de la operación y transacción de reporte. | 20 | A | Número de la factura o documento equivalente que evidencia el precio reportado en el campo 12. En caso de que la transacción se haya realizado a través de un contrato, se debe reportar con el número de este. | SÍ |
| 16 | NIT del actor con quien se realizó la operación y transacción de reporte, al precio máximo. | 12 | N | Número de Identificación Tributaria (NIT), sin incluir el código de verificación, del actor con el cual se realizó la operación y transacción para el mes de reporte en la factura de mayor valor. | SÍ |
| 17 | Número de factura o documento equivalente del precio máximo unitario de la operación y transacción de reporte. | 20 | A | Número de la factura o documento equivalente que evidencia el precio reportado en el campo 13. En caso de que la transacción se haya realizado a través de un contrato, se debe reportar con el número de este. | SÍ |
Aclaraciones:
1. En el caso en que se registren descuentos comerciales en las facturas del mes, para la operación y transacción respectiva, únicamente se debe reportar los precios finales que se encuentran facturados.
2. Las unidades bonificadas no deberán considerarse para el reporte.
3. Se aclara que las operaciones en las que la factura no indique el valor del dispositivo médico, se debe reportar el número de contrato o documento equivalente donde se evidencie el dispositivo médico objeto de reporte y su respectivo precio.
4. Con el fin de que las auditorías realizadas por los entes de control puedan dar cuenta de los precios reportados para los dispositivos médicos que sean comercializados o dispensados por concepto de contratos, el actor obligado a reportar deberá contar con el respectivo soporte documental que indique el precio de referencia por valor unitario de cada dispositivo médico reportado.
Las facturas asociadas a operaciones de maquila o traspasos de inventario no deben ser consideradas en las facturas objeto de reporte.
Características de los archivos planos
Los archivos deben ser tipo texto y cumplir con las siguientes especificaciones técnicas:
2.1. En el anexo técnico de los archivos, el tipo de dato corresponde a los siguientes:
A-Alfanumérico N-Numérico D-decimal F-Fecha T-Texto con caracteres especiales.
2.2. Todos los datos deben ser grabados como texto en archivos planos de formato ANSI, con extensión.txt
2.3. Los nombres de archivos y los datos de estos deben ser grabados en letras MAYÚSCULAS, sin caracteres especiales y sin tildes.
2.4. El separador de campos debe ser pipe (|) y debe ser usado exclusivamente para este fin. Los campos que corresponden a descripciones no deben incluir el carácter especial pipe (|).
2.5. Cuando dentro de un archivo de datos se definan campos que no son obligatorios y que no sean reportados, este campo no llevará ningún valor, es decir, debe ir vacío y reportarse en el archivo entre dos pipes, por ejemplo, si entre el dato1 y el dato3, el dato2 está vacío se reportará así: dato1||dato3.
2.6. Ningún dato en el campo debe venir encerrado entre comillas (““) ni ningún otro carácter especial.
2.7. Los campos numéricos deben venir sin ningún formato de valor ni separación de miles. Para los campos que se permita valores decimales, se debe usar el punto (.) como separador de decimales.
2.8. Los campos de tipo fecha deben venir en formato AAAA-MM-DD incluido el carácter guion, a excepción de las fechas que hacen parte del nombre de los archivos.
2.9. Las longitudes de campos definidas en los registros de control y detalle de este anexo técnico se deben entender como el tamaño máximo del campo, es decir, que los datos pueden tener una longitud menor al tamaño máximo.
2. 10. Los valores registrados en los archivos planos no deben tener ninguna justificación, por lo tanto, no se les debe completar con ceros ni espacios.
2. 11. Tener en cuenta que cuando los códigos traen CEROS, estos no pueden ser remplazados por la vocal 'O' la cual es un carácter diferente a cero.
2. 12. Los archivos planos no deben traer ningún carácter especial de fin de archivo ni de final de registro. Se utiliza el ENTER como fin de registro.
2. 13. Los archivos deben estar firmados digitalmente.
2. Plataforma para el envío de archivos.
El Ministerio de Salud y Protección Social dispondrá de la Plataforma de Intercambio de Información, PISIS, del Sistema Integral de Información de la Protección Social, Sispro, para que los actores reporten la información desde sus instalaciones. Si el reportante aún no tiene usuario debe solicitarlo previo registro de su entidad en el sitio web del Sispro.
Registrar entidad:
http://web.sispro.gov.co/WebPublico/Entidades/RegistrarEntidad.aspx
Registrar solicitud de usuario:
http://web. sispro.gov.co/Seguridad/Cliente/Web/RegistroSolicitudes.aspx
Control de calidad de los datos.
La Plataforma Pisis recibe los archivos conformados según la estructura del presente Anexo Técnico determinado en este acto administrativo y realiza el proceso de validación, así:
Primera validación: corresponde a la revisión de la estructura de los datos y se informa el estado de la recepción al reportante.
Segunda validación: Una vez realizada en forma exitosa la primera validación se realiza el control de calidad de contenido en el aplicativo misional y se informa al reportante el resultado.
Se entiende cumplida la obligación de este reporte una vez la segunda validación sea exitosa.
Mesa de ayuda.
Con el propósito de brindar ayuda técnica para el reporte de los archivos, transporte de datos y demás temas relacionados, el Ministerio de Salud y Protección Social tiene dispuesta la mesa de ayuda para uso de la plataforma PISIS. Los datos de contacto se encuentran en el siguiente enlace:
http://www.sispro.gov.co/recursosapp/Pages/Mesa_Ayuda.aspx
Adicionalmente, se dispone de documentación para el uso de la plataforma PISIS en el siguiente enlace:
http://web.sispro.gov.co/WebPublico/Soporte/FAQ/FAQ.aspx
Tratamiento de la información.
Las entidades y actores que participen en el flujo y consolidación de la información, se hacen responsables de la privacidad, seguridad, confidencialidad y veracidad de la información suministrada y sobre los datos a los cuales tiene acceso.
Seguridad de la información.
Para garantizar la seguridad y veracidad de la información reportada, las entidades deben enviar los archivos firmados digitalmente, lo cual protege los archivos garantizando su confidencialidad, integridad y no repudio. Para firmar digitalmente los archivos, se debe usar un certificado digital emitido por una entidad certificadora abierta aprobada por la entidad competente.
Las notas de vigencia, concordancias, notas del editor, forma de presentación y disposición de la compilación están protegidas por las normas sobre derecho de autor. En relación con estos valores jurídicos agregados, se encuentra prohibido por la normativa vigente su aprovechamiento en publicaciones similares y con fines comerciales, incluidas -pero no únicamente- la copia, adaptación, transformación, reproducción, utilización y divulgación masiva, así como todo otro uso prohibido expresamente por la normativa sobre derechos de autor, que sea contrario a la normativa sobre promoción de la competencia o que requiera autorización expresa y escrita de los autores y/o de los titulares de los derechos de autor. En caso de duda o solicitud de autorización puede comunicarse al teléfono 617-0729 en Bogotá, extensión 101. El ingreso a la página supone la aceptación sobre las normas de uso de la información aquí contenida.